![]()
Arthropods/COI
This is executable step-by-step pipeline for COI amplicon data from Illumina sequencing machine. The bioinformatic workflow results in amplicon sequence variants (ASVs) and well as operational taxonomic units (OTUs).
Note
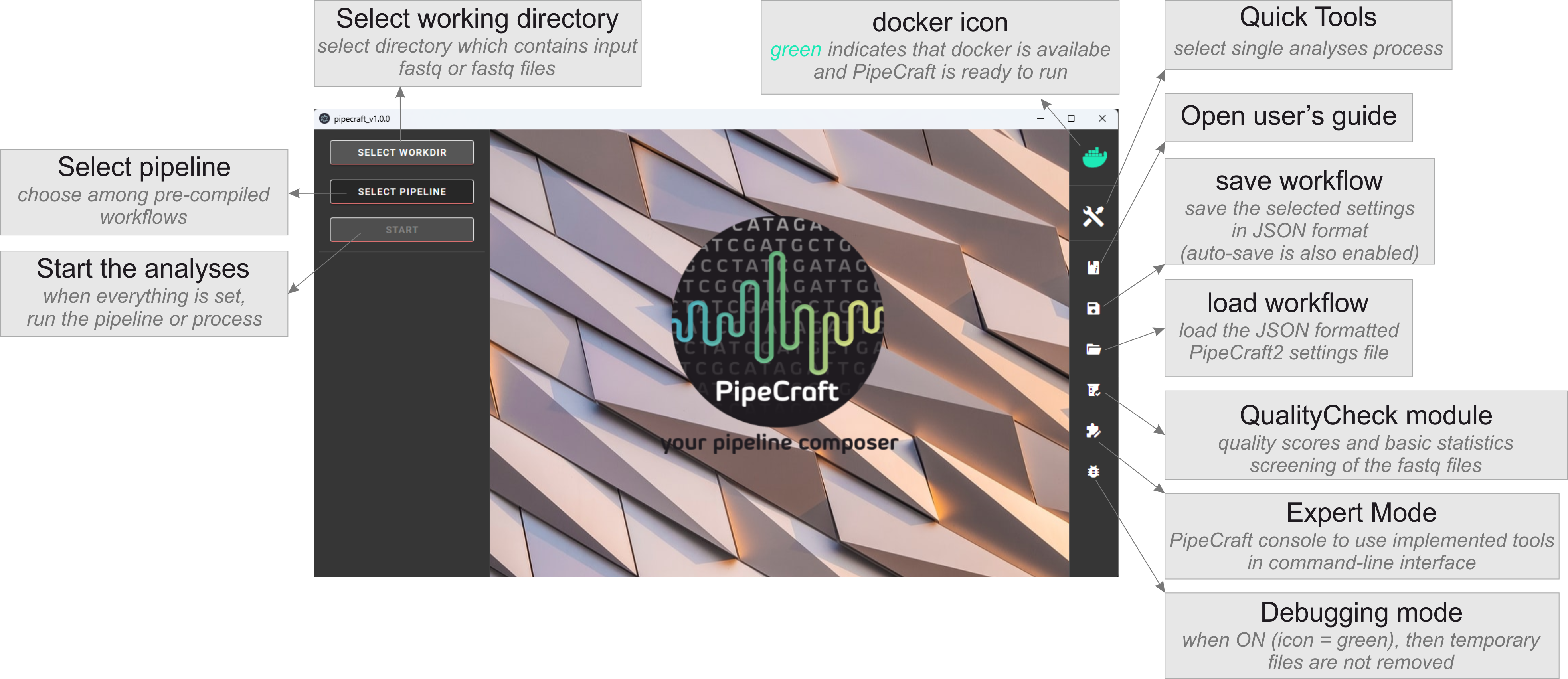
The full bioinformatics workflow can be automatically run through PipeCraft2, a software package which represents a graphical user interface wrapper for all the bioinformatic steps below. See the example workflow for COI.
Citation of the pipeline
When using this pipeline, please cite as: Anslan, S, Bahram, M, Hiiesalu, I, Tedersoo, L. PipeCraft: Flexible open-source toolkit for bioinformatics analysis of custom high-throughput amplicon sequencing data. Mol Ecol Resour. 2017; 17: e234-e240. https://doi.org/10.1111/1755-0998.12692
Also, please cite the original resources of the wrapped software.
Dependencies
Process |
Software |
Version |
|---|---|---|
cutadapt |
4.9 |
|
DADA2 |
1.30 |
|
DADA2 |
1.30 |
|
DADA2 |
1.30 |
|
DADA2 |
1.30 |
|
UNCROSS2 |
||
DADA2 |
1.30 |
|
vsearch |
2.28.1 |
|
R |
||
metaMATE |
0.4.3 |
|
vsearch |
2.28.1 |
|
BLAST |
2.12.0+ |
|
LULU |
0.1.0+ |
*only applicable when there are multiple sequencing runs per study.
Note
All the dependencies/software of the pipeline are available on a Docker image.
docker pull pipecraft/bioscanflow:2
# run docker
# specify the files location with -v flag ($PWD = the current working directory)
docker run -i --tty -v $PWD/:/Files pipecraft/bioscanflow:2
# inside the container, the files are accessible in the /Files dir
cd Files
# checking if cutadapt is available
cutadapt -h
# ready to run the pipe as below ...
## make sure that via the shared folder (-v) path you have access also to the reference databases.
# pull the docker image and save it as a singularity image
apptainer pull --name bioscanflow_2.sif docker://pipecraft/bioscanflow:2
# test if eg cutadapt is available
apptainer exec bioscanflow_2.sif cutadapt -h
# run the script via singularity image
apptainer exec --bind $PWD:/Files bioscanflow_2.sif /Files/run_pipe.sh
Data structure
Multiple sequencing runs
Important
When aiming to combine samples from multiple sequencing runs, then follow the below directory structure
Directory structure:
Note
Fastq files with the same name will be considered as the same sample and will be merged in the “Merge sequencing runs” step.
Single sequencing run
Remove primers
Note
Here, assuming that all sequences are in 5’-3’ orientation! (3’-5’ orient sequences will be discarded with this workflow)
Important
1 #!/bin/bash
2 ## workflow to remove primers with cutadapt
3
4 # specify the identifier string for the R1 files
5 read_R1="_R1"
6
7 # specify primers
8 fwd_primer=$"CCHGAYATRGCHTTYCCHCG" # this is the forward primer BF3
9 rev_primer=$"CDGGRTGNCCRAARAAYCA" # this is the reverse primer BR2
10
11 # specify primers
12 #fwd_primer=$"GGWACWRGWTGRACWITITAYCCYCC" # this is the forward primer mICOIintF-XT
13 #rev_primer=$"TAIACYTCIGGRTGICCRAARAAYCA" # this is the reverse primer jgHCO2198
14
15 # specify primers
16 #fwd_primer=$"GGDACWGGWTGAACWGTWTAYCCHCC" # this is the forward primer FwhF2
17 #rev_primer=$"GTRATWGCHCCDGCTARWACWGG" # this is the reverse primer FwhR2n
18
19 # edit primer trimming settings
20 mismatches="2" # Numer of allowed mismatches in primer string search;
21 # if set as 1, then allow 1 mismatch;
22 # if set as 0.1, then allow mismatch in 10% of the bases.
23 overlap="22" # The minimum overlap length. Keep it nearly as high
24 # as the primer length to avoid short random matches.
25 pair_filter="any" # Option 'any' discards a read pair if primers are not found in
26 # either of the read pairs (R1 and R2).
27 # Option 'both' keeps the read pair if a primer is found in
28 # at least one of the read pairs.
29 ##
30 # get the reverse complementary of the primers
31 # needed when the amplicon length is shorter than the sequencing cycle
32 fwd_primer_rc=$(echo $fwd_primer | rev | tr "ACGTRYKMBDHV" "TGCAYRMKVHDB")
33 rev_primer_rc=$(echo $rev_primer | rev | tr "ACGTRYKMBDHV" "TGCAYRMKVHDB")
34
35 # get directory names if working with multiple sequencing runs
36 # in that case, my working folder = /multiRunDir (see dir structure above)
37 DIRS=$(ls -d *) # -> sequencing_set01 sequencing_set02 sequencing_set03
38
39 for sequencing_run in $DIRS; do
40 printf "\nWorking with $sequencing_run \n"
41 cd $sequencing_run
42 #-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#
43 # make output dirs
44 mkdir -p primersCut_out
45 mkdir -p primersCut_out/untrimmed
46
47 ### Clip primers with cutadapt
48 for inputR1 in *$read_R1*; do
49 inputR2=$(echo $inputR1 | sed -e 's/R1/R2/')
50 cutadapt --quiet \
51 -e $mismatches \
52 --minimum-length 32 \
53 --overlap $overlap \
54 --no-indels \
55 --cores=0 \
56 --untrimmed-output primersCut_out/untrimmed/$inputR1 \
57 --untrimmed-paired-output primersCut_out/untrimmed/$inputR2 \
58 --pair-filter=$pair_filter \
59 -g $fwd_primer \
60 -a $fwd_primer...$rev_primer_rc";optional" \
61 -G $rev_primer \
62 -A $rev_primer...$fwd_primer_rc";optional" \
63 -o primersCut_out/$inputR1 \
64 -p primersCut_out/$inputR2 \
65 $inputR1 $inputR2
66 done
67 #-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#
68 cd ..
69 done
Quality filtering
1 #!/usr/bin/Rscript
2 ## workflow to perform quality filtering within DADA2
3
4 #load dada2 library
5 library('dada2')
6
7 # specify the identifier string for the R1 files
8 read_R1 = "_R1"
9
10 # get the identifier string for the R2 files
11 read_R2 = gsub("R1", "R2", read_R1)
12
13 # capturing the directory structure when working with multiple runs
14 wd = getwd() # -> wd is "~/multiRunDir"
15 dirs = list.dirs(recursive = FALSE)
16 for (i in 1:length(dirs)) {
17 if(length(dirs) > 1) {
18 setwd(dirs[i])
19 print(paste0("Working with ", dirs[i]))
20 #-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#
21 # output path
22 path_results = "qualFiltered_out"
23 # input and output file paths
24 R1s = sort(list.files("primersCut_out", pattern = read_R1, full.names = TRUE))
25 R2s = sort(list.files("primersCut_out", pattern = read_R2, full.names = TRUE))
26 #sample names
27 sample_names = sapply(strsplit(basename(R1s), read_R1), `[`, 1)
28
29 # filtered files path
30 filtR1 = file.path(path_results, paste0(sample_names, ".R1.", "fastq.gz"))
31 filtR2 = file.path(path_results, paste0(sample_names, ".R2.", "fastq.gz"))
32 names(filtR1) = sample_names
33 names(filtR2) = sample_names
34
35 #quality filtering
36 qfilt = filterAndTrim(R1s, filtR1, R2s, filtR2,
37 maxN = 0, # max number of allowed N bases.
38 maxEE = c(2, 2), # max error rate per R1 and R2 read, respectively.
39 truncQ = 2, # truncate reads at the first instance of a quality score less than or equal to specified value.
40 truncLen = c(0, 0), # truncate reads after specified length for R1 and R2 reads, respectively.
41 maxLen = 600, # discard reads longer than specified.
42 minLen = 100, # discard reads shorter than specified.
43 minQ = 2, # discard reads (after truncation) that contain a quality score below specified value.
44 matchIDs = TRUE, # output paired-end reads with matching IDs (for merging).
45 compress = TRUE, # gzip the output
46 multithread = TRUE) # use multiple threads
47 saveRDS(qfilt, file.path(path_results, "qfilt_reads.rds"))
48
49 # make sequence count report
50 seq_count = cbind(qfilt)
51 colnames(seq_count) = c("input", "qualFiltered")
52 seq_count = as.data.frame(seq_count)
53 seq_count$sample = sample_names
54 # reorder columns
55 seq_count = seq_count[, c("sample", "input", "qualFiltered")]
56 write.csv(seq_count, file.path(path_results, "seq_count_summary.csv"),
57 row.names = FALSE, quote = FALSE)
58
59 # save filtered R objects for denoising and merging (below)
60 filtR1 = sort(list.files(path_results, pattern = ".R1.fastq.gz", full.names = TRUE))
61 filtR2 = sort(list.files(path_results, pattern = ".R2.fastq.gz", full.names = TRUE))
62 sample_names = sapply(strsplit(basename(filtR1), ".R1.fastq.gz"), `[`, 1)
63 saveRDS(filtR1, file.path(path_results, "filtR1.rds"))
64 saveRDS(filtR2, file.path(path_results, "filtR2.rds"))
65 saveRDS(sample_names, file.path(path_results, "sample_names.rds"))
66 #-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#
67 #set working directory back to "/multiRunDir"
68 setwd(wd)
69 i = i + 1
70 }
71 }
Denoise and merge paired-end reads
1 #!/usr/bin/Rscript
2 ## workflow to perform DADA2 denoising and merging
3
4 # load dada2 library
5 library('dada2')
6
7 # capturing the directory structure when working with multiple runs
8 wd = getwd() # -> wd is "~/multiRunDir"
9 dirs = list.dirs(recursive = FALSE)
10 for (i in 1:length(dirs)) {
11 if(length(dirs) > 1) {
12 setwd(dirs[i])
13 print(paste0("Working with ", dirs[i]))
14 #-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#
15 #load quality filtered files
16 filtR1 = readRDS("qualFiltered_out/filtR1.rds")
17 filtR2 = readRDS("qualFiltered_out/filtR2.rds")
18 qfilt = readRDS("qualFiltered_out/qfilt_reads.rds")
19 sample_names = readRDS("qualFiltered_out/sample_names.rds")
20 cat("\n sample names = ", sample_names, "\n ")
21 names(filtR1) = sample_names
22 names(filtR2) = sample_names
23
24 # create output dir
25 path_results = "denoised_merged"
26 dir.create(path_results, showWarnings = FALSE)
27
28 print("# Denoising ...")
29 # learn the error rates
30 errF = learnErrors(filtR1, multithread = TRUE)
31 errR = learnErrors(filtR2, multithread = TRUE)
32
33 # make error rate figures
34 pdf(file.path(path_results, "Error_rates_R1.pdf"))
35 print( plotErrors(errF) )
36 dev.off()
37 pdf(file.path(path_results, "Error_rates_R2.pdf"))
38 print( plotErrors(errR) )
39 dev.off()
40
41 # Sample inference and merger of paired-end reads
42 mergers = vector("list", length(sample_names))
43 names(mergers) = sample_names
44 for(sample in sample_names) {
45 cat("\n -- Processing:", sample, "\n")
46 derepF = derepFastq(filtR1[[sample]])
47 ddF = dada(derepF, err = errF, multithread = TRUE)
48 derepR = derepFastq(filtR2[[sample]])
49 ddR = dada(derepR, err = errR, multithread = TRUE)
50 merger = mergePairs(ddF, derepF, ddR, derepR)
51 mergers[[sample]] = merger
52 }
53 rm(derepF); rm(derepR)
54 gc()
55 saveRDS(mergers, (file.path(path_results, "mergers.rds")))
56
57 # make sequence table
58 ASV_tab = makeSequenceTable(mergers)
59 #write RDS object
60 saveRDS(ASV_tab, (file.path(path_results, "rawASV_table.rds")))
61
62 # make sequence count report
63 getN = function(x) sum(getUniques(x))
64 #remove 0 seqs samples from qfilt statistics
65 row_sub = apply(qfilt, 1, function(row) all(row !=0 ))
66 qfilt = qfilt[row_sub, ]
67 seq_count = cbind(qfilt, sapply(mergers, getN))
68 colnames(seq_count) = c("input", "qualFiltered", "denoised_and_merged")
69 rownames(seq_count) = sample_names
70 write.csv(seq_count, file.path(path_results, "seq_count_summary.csv"),
71 row.names = TRUE, quote = FALSE)
72 #-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#
73 print("--------")
74 setwd(wd)
75 i = i + 1
76 }
77 }
Chimera filtering
1 #!/usr/bin/Rscript
2 ## workflow to perform chimera filtering within DADA2
3
4 # load libraries
5 library('dada2')
6 library('openssl')
7
8 # chimera filtering method
9 method = "consensus"
10
11 # collapse ASVs that have no mismatshes or internal indels (identical up to shifts and/or length)
12 collapseNoMismatch = "true" #true/false
13
14 # capturing the directory structure when working with multiple runs
15 wd = getwd() # -> wd is "~/multiRunDir"
16 dirs = list.dirs(recursive = FALSE)
17 for (i in 1:length(dirs)) {
18 if(length(dirs) > 1) {
19 setwd(dirs[i])
20 print(paste0("Working with ", dirs[i]))
21 #-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#
22 # load denoised and merged ASVs
23 rawASV_table = readRDS("denoised_merged/rawASV_table.rds")
24 # create output dir
25 path_results="ASV_table"
26 dir.create(path_results, showWarnings = FALSE)
27 # Remove chimeras
28 print("Removing chimeric ASVs ...")
29 chim_filt = removeBimeraDenovo(
30 rawASV_table, method = method,
31 multithread = TRUE,
32 verbose = TRUE)
33 saveRDS(chim_filt, "ASV_table/chim_filt.rds")
34
35 ### format and save ASV table and ASVs.fasta files
36 # sequence headers
37 asv_seqs = colnames(chim_filt)
38 asv_headers = openssl::sha1(asv_seqs)
39 # transpose sequence table
40 tchim_filt = t(chim_filt)
41 # add sequences to 1st column
42 tchim_filt = cbind(row.names(tchim_filt), tchim_filt)
43 colnames(tchim_filt)[1] = "Sequence"
44 # row names as sequence headers
45 row.names(tchim_filt) = asv_headers
46 # write ASVs.fasta to path_results
47 asv_fasta = c(rbind(paste(">", asv_headers, sep=""), asv_seqs))
48 write(asv_fasta, file.path(path_results, "ASVs.fasta"))
49 # write ASVs table to path_results
50 write.table(tchim_filt, file.path(path_results, "ASV_table.txt"),
51 sep = "\t", col.names = NA,
52 row.names = TRUE, quote = FALSE)
53
54 ### collapse ASVs that have no mismatshes or internal indels
55 # (identical up to shifts and/or length)
56 if (collapseNoMismatch == "true") {
57 print("Collapsing identical ASVs ...")
58 ASV_tab_collapsed = collapseNoMismatch(chim_filt,
59 minOverlap = 20, orderBy = "abundance",
60 identicalOnly = FALSE, vec = TRUE,
61 band = -1, verbose = TRUE)
62 saveRDS(ASV_tab_collapsed, file.path(path_results, "ASV_table_collapsed.rds"))
63
64 ### format and save ASV table and ASVs.fasta files
65 # sequence headers
66 asv_seqs = colnames(ASV_tab_collapsed)
67 asv_headers = openssl::sha1(asv_seqs)
68 # transpose sequence table
69 tASV_tab_collapsed = t(ASV_tab_collapsed)
70 # add sequences to 1st column
71 tASV_tab_collapsed = cbind(row.names(tASV_tab_collapsed), tASV_tab_collapsed)
72 colnames(tASV_tab_collapsed)[1] = "Sequence"
73 #row names as sequence headers
74 row.names(tASV_tab_collapsed) = asv_headers
75 # write ASVs.fasta to path_results
76 asv_fasta = c(rbind(paste(">", asv_headers, sep=""), asv_seqs))
77 write(asv_fasta, file.path(path_results, "ASVs_collapsed.fasta"))
78 # write ASVs table to path_results
79 write.table(tASV_tab_collapsed, file.path(path_results, "ASVs_table_collapsed.txt"),
80 sep = "\t", col.names = NA, row.names = TRUE, quote = FALSE)
81
82 # print summary
83 print(paste0("Output = ", length(colnames(ASV_tab_collapsed)),
84 " chimera filtered (+collapsed) ASVs, with a total of ",
85 sum(rowSums(ASV_tab_collapsed)),
86 " sequences."))
87 print("--------")
88 } else {
89 # print summary
90 print(paste0("Output = ", length(colnames(chim_filt)),
91 " chimera filtered ASVs, with a total of ",
92 sum(rowSums(chim_filt)),
93 " sequences."))
94 print("--------")
95 }
96 #-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#
97 setwd(wd)
98 i = i + 1
99 }
100 }
Remove tag-jumps
1#!/usr/bin/Rscript
2## Script to perform tag-jump removal; (C) Vladimir Mikryukov,
3 # edit, Sten Anslan
4
5 # load libraries
6 library(data.table)
7
8 # set parameters
9 set_f = 0.03 # f-parameter of UNCROSS (e.g., 0.03)
10 set_p = 1 # p-parameter (e.g., 1.0)
11
12 # output dir
13 path_results="ASV_table"
14
15 # capturing the directory structure when working with multiple runs
16 wd = getwd() # -> wd is "~/multiRunDir"
17 dirs = list.dirs(recursive = FALSE)
18 for (i in 1:length(dirs)) {
19 if(length(dirs) > 1) {
20 setwd(dirs[i])
21 print(paste0("Working with ", dirs[i]))
22 #-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#
23 # load ASV table
24 # loading ASV_table_collapsed if collapseNoMismatch was "true" (above)
25 if (file.exists("ASV_table/ASV_table_collapsed.rds") == TRUE) {
26 tab = readRDS("ASV_table/ASV_table_collapsed.rds")
27 cat("input table = ASV_table/ASV_table_collapsed.rds\n")
28 } else { # loading chimera filtered ASV table
29 tab = readRDS("ASV_table/chim_filt.rds")
30 cat("input table = ASV_table/chim_filt.rds\n")
31 }
32
33 # format ASV table
34 ASVTABW = as.data.table(t(tab), keep.rownames = TRUE)
35 colnames(ASVTABW)[1] = "ASV"
36 # convert to long format
37 ASVTAB = melt(data = ASVTABW, id.vars = "ASV",
38 variable.name = "SampleID", value.name = "Abundance")
39 # remove zero-OTUs
40 ASVTAB = ASVTAB[ Abundance > 0 ]
41 # estimate total abundance of sequence per plate
42 ASVTAB[ , Total := sum(Abundance, na.rm = TRUE), by = "ASV" ]
43
44 ## UNCROSS score
45 uncross_score = function(x, N, n, f = 0.01, tmin = 0.1, p = 1){
46 z = f * N / n # Expected treshold
47 sc = 2 / (1 + exp(x/z)^p) # t-score
48 res = data.table(Score = sc, TagJump = sc >= tmin)
49 return(res)
50 }
51
52 # esimate UNCROSS score
53 cat(" estimating UNCROSS score\n")
54 ASVTAB = cbind(
55 ASVTAB,
56 uncross_score(
57 x = ASVTAB$Abundance,
58 N = ASVTAB$Total,
59 n = length(unique(ASVTAB$SampleID)),
60 f = as.numeric(set_f),
61 p = as.numeric(set_p)
62 )
63 )
64 cat(" number of tag-jumps: ", sum(ASVTAB$TagJump, na.rm = TRUE), "\n")
65
66 # tag-jump stats
67 TJ = data.table(
68 Total_reads = sum(ASVTAB$Abundance),
69 Number_of_TagJump_Events = sum(ASVTAB$TagJump),
70 TagJump_reads = sum(ASVTAB[ TagJump == TRUE ]$Abundance, na.rm = T))
71
72 TJ$ReadPercent_removed = with(TJ, (TagJump_reads / Total_reads * 100))
73 fwrite(x = TJ, file = "ASV_table/TagJump_stats.txt", sep = "\t")
74
75 # prepare ASV tables, remove tag-jumps
76 ASVTAB = ASVTAB[ TagJump == FALSE ]
77 # convert to wide format
78 RES = dcast(data = ASVTAB,
79 formula = ASV ~ SampleID,
80 value.var = "Abundance", fill = 0)
81 # sort rows (by total abundance)
82 clz = colnames(RES)[-1]
83 otu_sums = rowSums(RES[, ..clz], na.rm = TRUE)
84 RES = RES[ order(otu_sums, decreasing = TRUE) ]
85
86 # output table that is compadible with dada2
87 output = as.matrix(RES, sep = "\t", header = TRUE, rownames = 1,
88 check.names = FALSE, quote = FALSE)
89 output = t(output)
90 saveRDS(output, ("ASV_table/ASV_table_TagJumpFiltered.rds"))
91
92 ### format and save ASV table and ASVs.fasta files
93 # sequence headers
94 asv_seqs = colnames(output)
95 asv_headers = openssl::sha1(asv_seqs)
96 # transpose sequence table
97 toutput = t(output)
98 # add sequences to 1st column
99 toutput = cbind(row.names(toutput), toutput)
100 colnames(toutput)[1] = "Sequence"
101 #row names as sequence headers
102 row.names(toutput) = asv_headers
103 # write ASVs.fasta to path_results
104 asv_fasta = c(rbind(paste(">", asv_headers, sep=""), asv_seqs))
105 write(asv_fasta, file.path(path_results, "ASVs_TagJumpFiltered.fasta"))
106 # write ASVs table to path_results
107 write.table(toutput, file.path(path_results, "ASV_table_TagJumpFiltered.txt"),
108 sep = "\t", col.names = NA, row.names = TRUE, quote = FALSE)
109
110 # print summary
111 print(paste0("Output = ", length(colnames(output)), " ASVs, with a total of ",
112 sum(rowSums(output)), " sequences."))
113
114 #-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#
115 print("--------")
116 setwd(wd)
117 i = i + 1
118 }
119 }
Merge sequencing runs
1 #!/usr/bin/Rscript
2 ## Merge sequencing runs, if working with multiple ones
3
4 # load libraries
5 library('dada2')
6
7 # after merging multiple ASV tables ...
8 # collapse ASVs that have no mismatshes or internal indels
9 collapseNoMismatch = "true" #true/false
10
11 # capturing the directory structure when working with multiple runs
12 wd = getwd() # -> wd is "~/multiRunDir"
13 dirs = list.dirs(recursive = FALSE)
14 tables = c()
15 # load tables from multiple sequencing runs (dirs)
16 for (i in 1:length(dirs)) {
17 if(length(dirs) > 1) {
18 setwd(dirs[i])
19 tables = append(tables, print(file.path(paste0(dirs[i], "/ASV_table"),
20 "ASV_table_TagJumpFiltered.rds")))
21 setwd(wd)
22 i = i + 1
23 }
24 }
25
26 # Merge multiple ASV tables
27 print("# Merging multiple ASV tables ...")
28 ASV_tables = lapply(tables, readRDS)
29 merged_table = mergeSequenceTables(tables = ASV_tables, repeats = "sum", tryRC = FALSE)
30
31 ### collapse ASVs that have no mismatshes or internal indels
32 if (collapseNoMismatch == "true") {
33 print("# Collapsing identical ASVs ...")
34 merged_table_collapsed = collapseNoMismatch(merged_table,
35 minOverlap = 20, orderBy = "abundance",
36 identicalOnly = FALSE, vec = TRUE,
37 band = -1, verbose = TRUE)
38 saveRDS(merged_table_collapsed, "merged_table_collapsed.rds")
39
40 ### format and save ASV table and ASVs.fasta files
41 # sequence headers
42 asv_seqs = colnames(merged_table_collapsed)
43 asv_headers = openssl::sha1(asv_seqs)
44 # transpose sequence table
45 tmerged_table_collapsed = t(merged_table_collapsed)
46 # add sequences to 1st column
47 tmerged_table_collapsed = cbind(row.names(tmerged_table_collapsed), tmerged_table_collapsed)
48 colnames(tmerged_table_collapsed)[1] = "Sequence"
49 #row names as sequence headers
50 row.names(tmerged_table_collapsed) = asv_headers
51 # write ASVs.fasta
52 asv_fasta = c(rbind(paste(">", asv_headers, sep=""), asv_seqs))
53 write(asv_fasta, "ASVs.merged_collapsed.fasta")
54 # write ASVs table
55 write.table(tmerged_table_collapsed, "ASV_table.merged_collapsed.txt",
56 sep = "\t", col.names = NA, row.names = TRUE, quote = FALSE)
57
58 # print summary
59 print(paste0("Output = ", length(colnames(merged_table_collapsed)),
60 " ASVs, with a total of ",
61 sum(rowSums(merged_table_collapsed)),
62 " sequences."))
63 } else {
64 saveRDS(merged_table, "merged_table.rds")
65 ### format and save ASV table and ASVs.fasta files
66 # sequence headers
67 asv_seqs = colnames(merged_table)
68 asv_headers = openssl::sha1(asv_seqs)
69 # transpose sequence table
70 tmerged_table = t(merged_table)
71 # add sequences to 1st column
72 tmerged_table = cbind(row.names(tmerged_table), tmerged_table)
73 colnames(tmerged_table)[1] = "Sequence"
74 #row names as sequence headers
75 row.names(tmerged_table) = asv_headers
76 # write ASVs.fasta to path_results
77 asv_fasta = c(rbind(paste(">", asv_headers, sep=""), asv_seqs))
78 write(asv_fasta, "ASVs.merged.fasta")
79 # write ASVs table to path_results
80 write.table(tmerged_table, "ASV_table.merged.txt",
81 sep = "\t", col.names = NA, row.names = TRUE, quote = FALSE)
82
83 # print summary
84 print(paste0("Output = ", length(colnames(merged_table)),
85 " ASVs, with a total of ",
86 sum(rowSums(merged_table)),
87 " sequences."))
88 }
Taxonomy assignment
1 #!/bin/bash
2
3 # download the BOLDistilled reference database
4 wget \
5 "https://us-sea-1.linodeobjects.com/boldistilled/sintax.zip"
6 # unzip the database
7 unzip sintax.zip
8
9 # specify reference database for SINTAX
10 reference_database="sintax/BOLDistilled_COI_Oct2025_SEQUENCES_sintax.fasta"
11 reference_database=$(realpath $reference_database) # get database names with full path
12
13 # specify input fasta file
14 cd ASV_table
15 ASV_fasta="ASVs_TagJumpFiltered.fasta"
16 ASV_fasta_tmp="ASVs_TagJumpFiltered_minmax.fasta"
17
18 # select by size to only retain ASVs that are within the range of expected variation. In this case we set it to 400 up to 430 bps
19 vsearch --fastx_filter $ASV_fasta \
20 --fastq_minlen 400 \
21 --fastq_maxlen 430 \
22 --fastaout $ASV_fasta_tmp
23
24 mv $ASV_fasta_tmp $ASV_fasta
25
26 # Run SINTAX classification
27 time vsearch --sintax $ASV_fasta \
28 --db $reference_database \
29 --tabbedout SINTAX.taxonomy.txt \
30 --sintax_cutoff 0.8 \
31 --threads 16
Get target taxa
1 #!/usr/bin/env Rscript
2 ### Filter dataset based on SINTAX results to include target taxa
3
4 # specify taxon and threshold
5 taxon="Animalia" # target taxonomic group(s);
6 # multiple groups should be from the same taxonomic level
7 # separator is "," (e.g., "Hymenoptera, Lepidoptera")
8 tax_level="kingdom" # allowed levels: kingdom | phylum | class | order | family | genus
9 threshold="0.8" # threshold for considering an ASV as a target taxon
10 class_threshold = 0.8 # threshold for class level identification
11
12 # specify the ASV table and ASVs.fasta file that would be filtered to include only target taxa
13 ASV_fasta = "ASVs_TagJumpFiltered.fasta"
14 ASV_table = "ASV_table_TagJumpFiltered.txt"
15
16 # specify the SINTAX-classifier output file (taxonomy file)
17 taxtab="SINTAX.taxonomy.txt"
18
19 #--------------------------------------#
20 library(stringr)
21 library(dplyr)
22 library(Biostrings)
23
24 # Function to parse SINTAX taxonomy format from vsearch output
25 parse_sintax = function(tax_string) {
26 # Initialize result with NAs
27 result = list(
28 kingdom = NA, kingdom_conf = 0,
29 phylum = NA, phylum_conf = 0,
30 class = NA, class_conf = 0,
31 order = NA, order_conf = 0,
32 family = NA, family_conf = 0,
33 genus = NA, genus_conf = 0,
34 species = NA, species_conf = 0
35 )
36
37 if (is.na(tax_string) || tax_string == "" || tax_string == "*") {
38 return(result)
39 }
40
41 # Split by comma
42 ranks = strsplit(tax_string, ",")[[1]]
43
44 for (rank in ranks) {
45 # Extract rank prefix (d:, k:, p:, c:, o:, f:, g:, s:)
46 if (grepl("^d:", rank)) {
47 # Domain (skip, not used)
48 next
49 } else if (grepl("^k:", rank)) {
50 # Kingdom
51 match = regmatches(rank, regexec("k:([^(]+)\\(([0-9.]+)\\)", rank))[[1]]
52 if (length(match) == 3) {
53 result$kingdom = match[2]
54 result$kingdom_conf = as.numeric(match[3])
55 }
56 } else if (grepl("^p:", rank)) {
57 # Phylum
58 match = regmatches(rank, regexec("p:([^(]+)\\(([0-9.]+)\\)", rank))[[1]]
59 if (length(match) == 3) {
60 result$phylum = match[2]
61 result$phylum_conf = as.numeric(match[3])
62 }
63 } else if (grepl("^c:", rank)) {
64 # Class
65 match = regmatches(rank, regexec("c:([^(]+)\\(([0-9.]+)\\)", rank))[[1]]
66 if (length(match) == 3) {
67 result$class = match[2]
68 result$class_conf = as.numeric(match[3])
69 }
70 } else if (grepl("^o:", rank)) {
71 # Order
72 match = regmatches(rank, regexec("o:([^(]+)\\(([0-9.]+)\\)", rank))[[1]]
73 if (length(match) == 3) {
74 result$order = match[2]
75 result$order_conf = as.numeric(match[3])
76 }
77 } else if (grepl("^f:", rank)) {
78 # Family
79 match = regmatches(rank, regexec("f:([^(]+)\\(([0-9.]+)\\)", rank))[[1]]
80 if (length(match) == 3) {
81 result$family = match[2]
82 result$family_conf = as.numeric(match[3])
83 }
84 } else if (grepl("^g:", rank)) {
85 # Genus
86 match = regmatches(rank, regexec("g:([^(]+)\\(([0-9.]+)\\)", rank))[[1]]
87 if (length(match) == 3) {
88 result$genus = match[2]
89 result$genus_conf = as.numeric(match[3])
90 }
91 } else if (grepl("^s:", rank)) {
92 # Species
93 match = regmatches(rank, regexec("s:([^(]+)\\(([0-9.]+)\\)", rank))[[1]]
94 if (length(match) == 3) {
95 result$species = match[2]
96 result$species_conf = as.numeric(match[3])
97 }
98 }
99 }
100
101 return(result)
102 }
103
104 # read ASV table
105 table = read.table(ASV_table, sep = "\t", check.names = F, header = T, row.names = 1)
106
107 # read SINTAX taxonomy table (vsearch --sintax output format)
108 # Format: ASV_ID \t taxonomy_string \t strand \t other_columns
109 tax_raw = read.table(taxtab, sep = "\t", check.names = F, header = F,
110 stringsAsFactors = F, quote = "", comment.char = "", fill = TRUE)
111
112 # Take first two columns only (ASV_ID and taxonomy)
113 tax_raw = tax_raw[, 1:2]
114 colnames(tax_raw) = c("ASV", "taxonomy")
115 rownames(tax_raw) = tax_raw$ASV
116
117 cat("\n Input =", nrow(tax_raw), "features.\n")
118
119 # Parse SINTAX taxonomy strings
120 tax_list = lapply(tax_raw$taxonomy, parse_sintax)
121 tax = do.call(rbind, lapply(tax_list, as.data.frame))
122 rownames(tax) = tax_raw$ASV
123
124 # taxon list
125 taxon_list = strsplit(taxon, ", ")[[1]]
126
127 ### extract only target-taxon ASVs from the 'raw' SINTAX results
128 tax_filtered = tax %>%
129 filter(.data[[tax_level]] %in% taxon_list)
130
131 cat("\n Found", nrow(tax_filtered), "ASVs matching", taxon, "at", tax_level, "level.\n")
132
133 ### Apply additional filter: class must be identified (confidence >= class_threshold)
134 tax_filtered = tax_filtered %>%
135 filter(class_conf >= class_threshold & !is.na(class))
136
137 cat(" After filtering for class identification (threshold >=", class_threshold, "):",
138 nrow(tax_filtered), "ASVs retained.\n")
139
140 ### change all tax ranks to "unclassified_*" when
141 # the confidence values is less than the specified threshold
142 # kingdom
143 tax_filtered = tax_filtered %>%
144 mutate(kingdom = ifelse(kingdom_conf < threshold | is.na(kingdom),
145 "unclassified_root", as.character(kingdom)))
146
147 # phylum
148 tax_filtered = tax_filtered %>%
149 mutate(phylum = ifelse(phylum_conf < threshold | is.na(phylum),
150 paste0("unclassified_", kingdom), as.character(phylum)))
151 tax_filtered$phylum = stringr::str_replace(tax_filtered$phylum, "unclassified_unclassified_",
152 "unclassified_")
153
154 # class
155 tax_filtered = tax_filtered %>%
156 mutate(class = ifelse(class_conf < threshold | is.na(class),
157 paste0("unclassified_", phylum), as.character(class)))
158 tax_filtered$class = stringr::str_replace(tax_filtered$class, "unclassified_unclassified_",
159 "unclassified_")
160
161 # order
162 tax_filtered = tax_filtered %>%
163 mutate(order = ifelse(order_conf < threshold | is.na(order),
164 paste0("unclassified_", class), as.character(order)))
165 tax_filtered$order = stringr::str_replace(tax_filtered$order, "unclassified_unclassified_",
166 "unclassified_")
167
168 # family
169 tax_filtered = tax_filtered %>%
170 mutate(family = ifelse(family_conf < threshold | is.na(family),
171 paste0("unclassified_", order), as.character(family)))
172 tax_filtered$family = stringr::str_replace(tax_filtered$family, "unclassified_unclassified_",
173 "unclassified_")
174
175 # genus
176 tax_filtered = tax_filtered %>%
177 mutate(genus = ifelse(genus_conf < threshold | is.na(genus),
178 paste0("unclassified_", family), as.character(genus)))
179 tax_filtered$genus = stringr::str_replace(tax_filtered$genus, "unclassified_unclassified_",
180 "unclassified_")
181
182 # species to genus_sp when the confidence values is < 0.9
183 tax_filtered = tax_filtered %>%
184 mutate(species = ifelse(species_conf < 0.9 | is.na(species),
185 paste0(genus, "_sp"), as.character(species)))
186
187 ### count occurrences of each taxon in df (SINTAX results)
188 count_taxa = function(df, taxa) {
189 sapply(taxa, function(taxon) sum(apply(df, 1, function(row) any(row == taxon))))
190 }
191 taxon_counts = count_taxa(tax_filtered, taxon_list)
192
193 # Check the counts
194 if (all(taxon_counts == 0)) {
195 print("ERROR: None of the specified taxa are present in the SINTAX results.")
196 } else {
197 if (any(taxon_counts == 0)) {
198 warning("One or more of the specified taxa are not present in the SINTAX results.")
199 }
200 cat("\n Taxon counts:\n")
201 print(taxon_counts)
202 }
203
204 ### extract only target-taxon ASVs from the 'threshold filtered' SINTAX results
205 tax_filtered_thresh = tax_filtered %>%
206 filter(.data[[tax_level]] %in% taxon_list)
207
208 # Remove confidence columns for output
209 tax_filtered_output = tax_filtered_thresh %>%
210 select(kingdom, phylum, class, order, family, genus, species)
211
212 # write filtered SINTAX taxonomy table
213 tax_filtered_output = cbind(ASV = rownames(tax_filtered_output), tax_filtered_output)
214 write.table(tax_filtered_output,
215 file = "SINTAX.taxonomy.filt.txt",
216 quote = F,
217 row.names = F,
218 sep = "\t")
219
220 ### filter the ASV table to match ASVs that were kept in the tax_filtered table
221 table_filt = table[rownames(table) %in% rownames(tax_filtered_thresh), ]
222
223 ### check ASV table; if 1st col is sequence, then remove it for metaMATE
224 if (colnames(table_filt)[1] == "Sequence") {
225 cat("\n;; 1st column was 'Sequence', removing this ... \n")
226 table_filt = table_filt[, -1]
227 }
228
229 # write filtered table
230 table_filt = cbind(ASV = rownames(table_filt), table_filt)
231 write.table(table_filt,
232 file = paste0(sub("\\.[^.]*$", "_tax_filt.txt", ASV_table)),
233 quote = F,
234 row.names = F,
235 sep = "\t")
236
237 # filter ASV_fasta
238 fasta = readDNAStringSet(ASV_fasta)
239 fasta.tax_filt = fasta[names(fasta) %in% rownames(table_filt)]
240
241 # write filtered ASV_fasta
242 writeXStringSet(fasta.tax_filt,
243 paste0(sub("\\.[^.]*$", "_tax_filt.fasta", ASV_fasta)),
244 width = max(width(fasta.tax_filt)))
If no pre-selection is preferred, then just remove “Sequence” column from the ASV table
1 # read ASV table
2 ASV_table = "ASV_table.txt"
3 table = read.table(ASV_table, sep = "\t", check.names = F, header = T, row.names = 1)
4
5 # check ASV table; if 1st col is sequence, then remove it for metaMATE
6 if (colnames(table)[1] == "Sequence") {
7 cat("## removing 'Sequence' column ... \n")
8 table = table[, -1]
9
10 # write filtered table
11 table_filt = cbind(ASV = rownames(table), table)
12 write.table(table_filt,
13 file = paste0(sub("\\.[^.]*$", ".noSeq.txt", ASV_table)),
14 quote = F,
15 row.names = F,
16 sep = "\t")
17
18 } else {
19 cat("## there was no 'Sequence' column; proceed with the current table ... \n")
20 }
Remove NUMTs
Important
1. metaMATE expects specifications file that states the filtering strategies. See more info here. Here, we will be using the metaMATE’s default specifications.txt file.
metaMATE requires a reference COI database to determine verified-authentic ASVs. Herein using BOLDistilled database.
— Download the latest BOLDistilled database here (click) —
If you have your own set of reference sequences, then use those; or merge those with the other databases (such as the above one) to extend the ref. database.
Check standard genetic codes here for genetic_code setting below.
1#!/bin/bash
2
3 # download the default specifications file,
4 # using this in metaMATE-find
5 wget "https://raw.githubusercontent.com/tjcreedy/metamate/main/specifications.txt"
6 # specify specifications file for metaMATE
7 specifications="specifications.txt"
8 specifications=$(realpath $specifications) # get full directory path
9
10
11 # download the BOLDistilled reference databse
12 wget \
13 "https://us-sea-1.linodeobjects.com/boldistilled/sintax.zip"
14 # unzip the database and edit name
15 unzip sintax.zip
16
17 # specify reference database for metaMATE
18 reference_database="sintax/BOLDistilled_COI_Oct2025_SEQUENCES_sintax.fasta"
19 reference_database=$(realpath $reference_database) # get full directory path
1 #!/bin/bash
2 ## run metaMATE-find
3
4 # cluster with 10% threshold
5 vsearch --cluster_fast ASVs_TagJumpFiltered_tax_filt.fasta --id 0.9 \
6 --uc ASVs_TagJumpFiltered_tax_filt_clustered.uc
7
8 # select only H & S
9 cat ASVs_TagJumpFiltered_tax_filt_clustered.uc | grep -v "^C" \
10 > ASVs_TagJumpFiltered_tax_filt_clustered_onlyHS.uc
11
12 # now extract the information to match the input requirements from metamate
13 awk -F'\t' 'BEGIN {OFS=","} {print $9, $2}' ASVs_TagJumpFiltered_tax_filt_clustered_onlyHS.uc \
14 > ASV_to_cluster_map.csv
1 #!/bin/bash
2 ## run metaMATE-find
3
4 ## go to the directory that hosts your ASVs.fasta and ASV table files.
5
6 # specify input ASVs table and fasta
7 ASV_table="ASV_table_TagJumpFiltered_tax_filt.txt" # make sure that the 2nd col is not "Sequence"
8 ASV_fasta="ASVs_TagJumpFiltered_tax_filt.fasta" # specify ASVs fasta file
9 taxgroups="ASV_to_cluster_map.csv" # comment out or change filename if sequence binning is done in another way
10
11 # specify variables
12 genetic_code="5" # the standard genetic code. 5 is invertebrate mitochondrial code
13 length="418" # the expected length of an amplicon
14 basesvariation="9" # allowed length variation (bp) from the expected length of an amplicon
15 taxgroups="undefined" # (optional); if sequence binning is to be performed on
16 # a per-taxon basis (as in specifications file)
17 # then specify the taxon grouping file
18 NA_abund_thresh="0.05" # verifiednonauthentic_retained_p < 0.05 (value from mateMATE results);
19 # the allowed abundance threshold of
20 # non-validated OTUs/ASVs in the filtered dataset.
21 abundance_filt="TRUE" # TRUE/FALSE; if FALSE, then NA_abund_thresh is ineffective,
22 # and no filtering is done based on the ASV abundances,
23 # i.e., filter only based on length, basesvariation and genetic_code.
24 # FALSE may be used when the seq-depth for the target taxa is low.
25 # If TRUE, then NA_abund_thresh will be applied.
26
27 ##
28
29 # check if taxgroups is specified, if not then this var is empty.
30 if [[ $taxgroups != "undefined" ]]; then
31 taxgroups=$"--taxgroups $taxgroups"
32 else
33 taxgroups=$""
34 fi
35
36 #output dir
37 output_dir=$"metamate_out"
38 echo "output_dir = $output_dir"
39 # remove old $output_dir if exists
40 if [[ -d $output_dir ]]; then
41 rm -rf $output_dir
42 fi
43
44 # if perfoming clade binning, then WARNING when processing more than 65,536 ASVs
45 ASVcount=$(grep -c "^>" $ASV_fasta)
46 if (( $ASVcount > 65536 )); then
47 printf '%s\n' "WARNING]: clade binning NOT performed,
48 because the input ASVs limit is 65,536 for that.
49 Current input has $ASVcount ASVs."
50 fi
51
52 # check abundance_filt;
53 # if FALSE then make new specifications file, that excludes abundance filtering
54 if [[ $abundance_filt == "FALSE" ]]; then
55 printf '%s\n' "[library; n; 0-1/2]" > specifications0.txt
56 specifications=$(realpath specifications0.txt)
57 fi
58
59 # quick check of the specifications file, has to contain "library" | "total" | "clade" | "taxon"
60 if ! grep -q -e "library" -e "total" -e "clade" -e "taxon" $specifications; then
61 printf '%s\n' "ERROR]: specifications file seems to be wrong.
62 Does not contain any of the terms (library, total, clade, taxon)."
63 fi
64
65 ### metaMATE-find
66 printf "# Running metaMATE-find\n"
67 metamate find \
68 --asvs $ASV_fasta \
69 --readmap $ASV_table \
70 --specification $specifications \
71 --references $reference_database \
72 --expectedlength $length \
73 --basesvariation $basesvariation \
74 --table $genetic_code \
75 --threads 8 \
76 --output $output_dir \
77 --overwrite $taxgroups \
78 --realign
79
80 # check for the presence of "metamate_out" dir and "resultcache" file (did metaMATE-find finish)
81 if [[ -d $output_dir ]] && [[ -e $output_dir/resultcache ]] && [[ -e $output_dir/results.csv ]]; then
82 printf '\n%s\n\n' "metaMATE-find finished, proceed"
83 # export variables for below script (Rscript)
84 if [[ $abundance_filt != "FALSE" ]]; then
85 printf '%s\n' "exporting NA_abund_thresh of $NA_abund_thresh for metaMATE-dump"
86 export NA_abund_thresh
87 else
88 export output_dir
89 export abundance_filt
90 fi
91 else
92 printf '%s\n' "ERROR]: cannot find the $output_dir (metaMATE-find output)
93 to start metaMATE-dump OR no authentic ASVs found??"
94 fi
1 #!/usr/bin/env Rscript
2
3 ## read results.csv
4 output_dir = Sys.getenv('output_dir') # = "metamate_out" as specified above
5 find_results = read.csv(file.path(output_dir, "results.csv"))
6
7 # get variables
8 abundance_filt = Sys.getenv('abundance_filt')
9
10 ## filter results if abundance_filt is FALSE
11 if (abundance_filt == "FALSE"){
12 result_index = "0" # get first result_index (library_n = 0)
13 write(result_index, file.path(output_dir, "selected_result_index.txt"))
14 }
15
16 ## filter results based on NA_abund_thresh
17 if (abundance_filt != "FALSE"){
18 NA_abund_thresh = as.numeric(Sys.getenv('NA_abund_thresh'))
19 filtered_data = find_results[
20 find_results$verifiednonauthentic_retained_p <= NA_abund_thresh, ]
21
22 # if no results correspond with the NA_abund_thresh, then get the next best
23 # else, just select the result_index that corresponds to
24 # NA_abund_thresh with highest accuracy_score
25 if (nrow(filtered_data) == 0) {
26 cat(
27 "\n no results correspond with the NA_abund_thresh of", NA_abund_thresh, ";
28 getting the next best setting\n"
29 )
30 next_best = min(find_results$verifiednonauthentic_retained_p)
31 filtered_data = find_results[
32 find_results$verifiednonauthentic_retained_p <= next_best, ]
33 # sort based on accuracy_score
34 sorted_filtered = filtered_data[order(-filtered_data$accuracy_score), ]
35 # get the result with the highest accuracy_score
36 metamate_selected_threshold = sorted_filtered[1,]
37 write.csv(metamate_selected_threshold, file.path(output_dir, "next_best_set.csv"),
38 quote = F)
39 # the result_index of the NA_abund_thresh with the highest accuracy_score
40 result_index = metamate_selected_threshold[,1]
41 write(result_index, file.path(output_dir, "selected_result_index.txt"))
42 } else {
43 # sort based on accuracy_score
44 sorted_filtered = filtered_data[order(-filtered_data$accuracy_score), ]
45 # get the result with the highest accuracy_score
46 metamate_selected_threshold = sorted_filtered[1,]
47 # the result_index of the NA_abund_thresh with the highest accuracy_score
48 result_index = metamate_selected_threshold[,1]
49 write(result_index, file.path(output_dir, "selected_result_index.txt"))
50 }
51 }
1 #!/bin/bash
2
3 ## metaMATE-dump
4 ASV_fasta=$(basename $ASV_fasta)
5
6 # read result_index
7 read -r result_index < $output_dir/selected_result_index.txt
8 printf '%s\n' " - selected result_index = $result_index"
9
10 # run metaMATE-dump
11 printf '%s\n' "# Running metaMATE-dump"
12 metamate dump \
13 --asvs $ASV_fasta \
14 --resultcache $output_dir/resultcache \
15 --output $output_dir/${ASV_fasta%.*}_metaMATE.filt \
16 --overwrite \
17 --resultindex $result_index
18
19 # generate a list of ASV IDs
20 seqkit seq -n $output_dir/${ASV_fasta%.*}_metaMATE.filt.fasta > \
21 $output_dir/${ASV_fasta%.*}_metaMATE.filt.list
22
23 # filter the ASV table; include only the ASVs that are in ${ASV_fasta%.*}_metaMATE.filt.list
24 awk -v var="$output_dir/${ASV_fasta%.*}" 'NR==1; NR>1 {print $0 | \
25 "grep -Fwf "var"_metaMATE.filt.list"}' $ASV_table > \
26 $output_dir/${ASV_table%.*}_metaMATE.filt.txt
27
28 # filter the sintax.taxonomy.filt.txt file to include only ASVs retained by metaMATE
29 awk -v var="$output_dir/${ASV_fasta%.*}" 'NR==1; NR>1 {print $0 | \
30 "grep -Fwf "var"_metaMATE.filt.list"}' sintax.taxonomy.filt.txt > \
31 $output_dir/sintax.taxonomy.metaMATE.filt.txt
32
33
34 # write discarded ASVs list
35 comm -3 <(sort <(seqkit seq -n $ASV_fasta)) \
36 <(sort $output_dir/${ASV_fasta%.*}_metaMATE.filt.list) \
37 > $output_dir/metaMATE.discarded.list
1 #!/bin/bash
2
3 # get discarded ASVs (sintax taxonomy list)
4 grep -Fwf $output_dir/metaMATE.discarded.list sintax.taxonomy.filt.txt \
5 > $output_dir/metaMATE.discarded.sintax.taxonomy.txt
6
7 # get the rescued ASVs that have GENUS level bootstrap value >= 0.9
8 awk -F'\t' '$26 >= 0.9' $output_dir/metaMATE.discarded.sintax.taxonomy.txt \
9 > $output_dir/rescued.txt
10
11 # check if rescued.txt exists and is not empty
12 if [[ -s $output_dir/rescued.txt ]]; then
13 # add the rescued ASVs to $output_dir/sintax.taxonomy.metaMATE.filt.txt
14 cat $output_dir/rescued.txt >> $output_dir/sintax.taxonomy.metaMATE.filt.txt
15
16 # add the rescued ASVs to $output_dir/${ASV_fasta%.*}_metaMATE.filt.fasta
17 seqkit grep -w 0 -f <(awk -F'\t' '{print $1}' $output_dir/rescued.txt) $ASV_fasta \
18 >> $output_dir/${ASV_fasta%.*}_metaMATE.filt.fasta
19
20 # add the rescued ASVs to $output_dir/${ASV_table%.*}_metaMATE.filt.txt
21 grep -wf <(awk -F'\t' '{print $1}' $output_dir/rescued.txt) $ASV_table \
22 >> $output_dir/${ASV_table%.*}_metaMATE.filt.txt
23
24 printf '%s\n' "Rescued $(wc -l < $output_dir/rescued.txt) ASVs"
25 else
26 printf '%s\n' "No ASVs to rescue"
27 fi
Note
Herein case, the final filtered data is ASV_table_tax_filt_metaMATE.filt.txt and ASVs_tax_filt_metaMATE.filt.fasta in the metamate_out directory.
The filtered SINTAX-classifier results (matching the ASVs in the latter files) is sintax.taxonomy.metaMATE.filt.txt in the metamate_out dir.
If deemed relevant, then you may proceed with the below workflow below that includes clustering ASVs to OTUs.
Clustering ASVs to OTUs
1 #!/usr/bin/env Rscript
2
3 # specify input ASVs table and fasta
4 ASV_table="ASV_table_tax_filt_metaMATE.filt.txt" # specify ASV table file
5 ASV_fasta="ASVs_tax_filt_metaMATE.filt.fasta" # specify ASVs fasta file
6
7 ################################
8 library(Biostrings)
9 # Read the ASV table
10 ASV_table = read.table(ASV_table, sep = "\t", check.names = F,
11 header = T, row.names = 1)
12
13 # add 'sum' column
14 ASV_table$sum = rowSums(ASV_table)
15 # make ASV_sums object
16 ASV_sums = setNames(ASV_table$sum, rownames(ASV_table))
17
18 # Read the FASTA file
19 ASV_fasta = readDNAStringSet(ASV_fasta)
20
21 # add ";size=*" to ASV_fasta
22 names(ASV_fasta) = sapply(names(ASV_fasta), function(header) {
23 paste0(header, ";size=", ASV_sums[header])
24 })
25 # write fasta file
26 writeXStringSet(ASV_fasta, "ASVs.size.fasta",
27 width = max(width(ASV_fasta)))
1 #!/bin/bash
2
3 # specify the clustering threshold
4 clustering_thresh="0.97"
5
6 # make output dir.
7 output_dir="OTU_table"
8 mkdir -p $output_dir
9 export output_dir
10
11 ### cluster ASVs using vsearch.
12 vsearch --cluster_fast ASVs.size.fasta \
13 --id $clustering_thresh \
14 --iddef 2 \
15 --sizein \
16 --xsize \
17 --fasta_width 0 \
18 --centroids $output_dir/OTUs.fasta \
19 --uc $output_dir/OTUs.uc
1 #!/usr/bin/Rscript
2
3 # specify input ASV table (the same one as for 'get the size of ASVs')
4 ASV_table="ASV_table_tax_filt_metaMATE.filt.txt"
5
6 # read output dir
7 output_dir = Sys.getenv('output_dir')
8
9 # read output from vsearch clustering (-uc OTU.uc)
10 inp_UC = file.path(output_dir, "OTUs.uc")
11 ################################
12 library(data.table)
13 # load input data - ASV table
14 ASV_table = fread(file = ASV_table, header = TRUE, sep = "\t")
15
16 ## Load input data - UC mapping file
17 UC = fread(file = inp_UC, header = FALSE, sep = "\t")
18 UC = UC[ V1 != "S" ]
19 UC[, ASV := tstrsplit(V9, ";", keep = 1) ]
20 UC[, OTU := tstrsplit(V10, ";", keep = 1) ]
21 UC[V1 == "C", OTU := ASV ]
22 UC = UC[, .(ASV, OTU)]
23
24 # convert ASV table to long format
25 ASV = melt(data = ASV_table,
26 id.vars = colnames(ASV_table)[1],
27 variable.name = "SampleID", value.name = "Abundance")
28 ASV = ASV[ Abundance > 0 ]
29 # add colnames, to make sure 1st is 'ASV'
30 colnames(ASV) = c("ASV", "SampleID", "Abundance")
31
32 # add OTU IDs
33 ASV = merge(x = ASV, y = UC, by = "ASV", all.x = TRUE)
34 # summarize
35 OTU = ASV[ , .(Abundance = sum(Abundance, na.rm = TRUE)),
36 by = c("SampleID", "OTU")]
37
38 # reshape OTU table to wide format
39 OTU_table = dcast(data = ASV,
40 formula = OTU ~ SampleID,
41 value.var = "Abundance",
42 fun.aggregate = sum, fill = 0)
43
44 # write OTU table
45 # OTU names correspond to most abundant ASV in an OTU
46 fwrite(x = OTU_table, file = file.path(output_dir,
47 "OTU_table.txt"), sep = "\t")
Post-clustering
Post-cluster OTUs with LULU to merge consistently co-occurring ‘daughter-OTUs’.
1 #!/bin/bash
2
3 # go to directrory that contains OTUs
4 cd $output_dir # 'OTU_table' in this case
5
6 # make blast database for post-clustering
7 makeblastdb -in OTUs.fasta -parse_seqids -dbtype nucl
8
9 # generate match list for post-clustering
10 blastn -db OTUs.fasta \
11 -outfmt '6 qseqid sseqid pident' \
12 -out match_list.txt \
13 -qcov_hsp_perc 75 \
14 -perc_identity 90 \
15 -query OTUs.fasta \
16 -num_threads 20
1 #!/usr/bin/Rscript
2
3 # specify minimum threshold of sequence similarity considering any OTU as an error of another
4 min_match = "90"
5
6 # specify OTU table
7 OTU_table="OTU_table.txt"
8
9 ################################
10 library(devtools)
11 # load OTU table and match list
12 otutable = read.table(OTU_table, header = T, row.names = 1, sep = "\t")
13 matchlist = read.table("match_list.txt")
14
15 curated_result = lulu::lulu(otutable, matchlist,
16 minimum_match = min_match)
17
18 # write post-clustered OTU table to file
19 curated_table = curated_result$curated_table
20 curated_table = cbind(OTU = rownames(curated_table), curated_table)
21 write.table(curated_table, file ="OTU_table_LULU.txt",
22 sep = "\t", row.names = F, quote = FALSE)
23 write.table(curated_result$discarded_otus,
24 file ="merged_units.lulu", col.names = FALSE, quote = FALSE)
Note
Note that if the sample names start with a number, then the output OTU table may contain “X” prefix in the sample names.
1 #!/bin/bash
2
3 # specify post-clustered table
4 OTU_table="OTU_table_LULU.txt"
5 # specify pre post-clustered OTUs fasta file
6 OTUs_fasta="OTUs.fasta"
7
8 # get matching OTUs
9 awk 'NR>1{print $1}' $OTU_table > OTUs_LULU.list
10 cat $OTUs_fasta | \
11 seqkit grep -w 0 -f OTUs_LULU.list > OTUs_LULU.fasta
12
13 # get matching sintax taxonomy results
14 head -n 1 ../sintax.taxonomy.metaMATE.filt.txt > sintax.taxonomy.txt
15 cat ../sintax.taxonomy.metaMATE.filt.txt | \
16 grep -wf OTUs_LULU.list >> sintax.taxonomy.txt
17
18 # remove unnecessary files
19 rm OTUs.fasta.n*
20
21 # move OTU_table two directories down
22 cd ..
23 mv $output_dir ../..
Note
The final OTUs data is OTU_table_LULU.txt and OTUs_LULU.fasta in the OTU_table directory.
The matching SINTAX taxonomy files are sintax.taxonomy.txt in the OTU_table directory.
We acknowledge CSC - IT Center for Science, Finland, for computational resources while building and testing this workflows.
![]()

![]()
![]()