Hierarchical Modelling of Species Communities
Hierarchical Modelling of Species Communities (HMSC) is a framework for Joint Species Distribution Modelling; a model-based approach for analyzing community ecological data (Ovaskainen et al.2017).
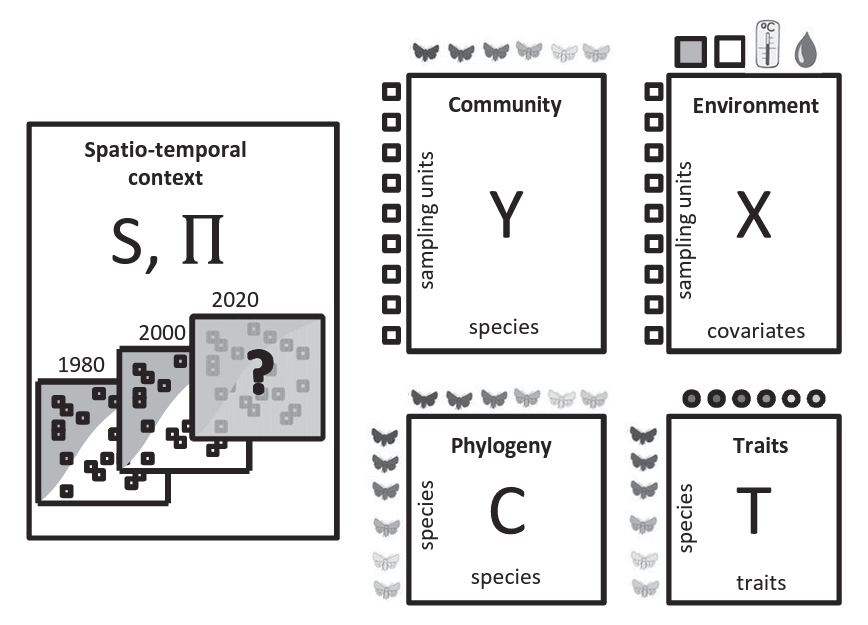
The input data for HMSC-analyses includes a matrix of species occurrences or abundances and a matrix of environmental covariates (sampling units as rows). Optionally additional input data include species traits and phylogeny, and information about the spatiotemporal context of the sampling design. HMSC yields inference both at species and community levels.
Starting point
The input for HMSC analysis consists of species/OTU community matrix (Y matrix) accompanied with the matrix of environmental covariates (X matrix), and optionally species traits (T) matrix and phylogeny (C) matrix.
Traits
Including the traits matrix helps to understand how species-specific traits influence the community assebly processes. Traits matrix may include data for example about body size, shape, feeding type, etc.
body_size |
shape |
trophic_guild |
|
OTU_01 |
2 |
square |
1 |
OTU_02 |
2 |
round |
1 |
OTU_03 |
0.1 |
round |
2 |
OTU_04 |
0.2 |
variable |
3 |
Phylogeny
Including the phylogeny matrix helps to understand if the species responses to the environmental covariates are phylogenetically structured, i.e., do similar species respond similarly.
The phylogeny matrix may be presented as Newick tree file. Alternatively, data on taxonomic ranks may used as a proxy of phylogenetic relatedness. In the latter case, the UNCLASSIFIED taxonomic ranks (that are common in the metabarcoding data) should be informative in a sense that not all e.g. family level unclassified OTUs would be considered as closely related because they have the ‘same label’. For example, the distance between unclassified OTUs could be calculated and then OTUs falling within the user defined distance threshold could be classified as various levels of pseudotaxa.
Example of the taxonomy table:
… |
Class |
Order |
Family |
Genus |
|
OTU_01 |
… |
Collembola |
Entomobryomorpha |
Entomobryidae |
Entomobrya |
OTU_02 |
… |
Collembola |
Entomobryomorpha |
Family_0032 |
Genus_001 |
OTU_03 |
… |
Collembola |
Entomobryomorpha |
Isotomidae |
Parisotoma |
OTU_04 |
… |
Collembola |
Entomobryomorpha |
Family_0032 |
Genus_022 |
Here, for example the sequences of OTU_02 and OTU_04 differ 9.8%; so we consider those are originating from different genera but likely from the same family.
Install HMSC
Install hmsc R package (if already not installed).
1#!/usr/bin/Rscript
2
3# install 'devtools'; if not yet installed
4install.packages("devtools")
5library(devtools)
6
7# install hmsc package
8install_github("hmsc-r/HMSC")
9
10# load hmsc
11library(Hmsc)
12
13# check the version
14packageVersion("Hmsc")
Select data
In this ‘select data’ section, we are assuming that our input Y and X matrixes are tab delimited text files where samples are in rows; and taxonomy table format follows the above example.
Here, we are also deciding if we want to proceed with the presence-absence or abundance (read count) community matrix.
Note
Before using to the full dataset, try fitting the model with a small subset for faster model testing and validation.
1#!/usr/bin/Rscript
2
3# input matrices file names; according to the above HMSC figure.
4 # here, expecting all files to be tab-delimited.
5Y_file = "OTU_table.txt" # Community; samples are rows
6X_file = "env_meta.txt" # Environment; samples are rows
7C_file = "taxonomy.txt" # Phylogeny (optional)
8 # (herein a 'pseudo-phylogeny' based on the
9 # assigned taxonomic ranks; species are rows)
10T_file = "" # Traits (optional)
11sp_prevalence = 100 # set a species prevalence threshold;
12 # meaning that perform HMSC for species
13 # in a Y-matrix, that occur >= 100 samples in this case.
14 #-------------------------------------------------------------------------------------------#
15
16 # load community matrix Y
17 Y = read.table(Y_file, sep = "\t",
18 check.names = F,
19 header = T,
20 row.names = 1)
21 # load metadata matrix X
22 X = read.table(X_file, sep = "\t",
23 check.names = F,
24 header = T,
25 row.names = 1)
26 # load taxonomy matrix C;
27 # so we can use it as a proxy for phylogeny
28 taxonomy = read.table(C_file, sep = "\t",
29 check.names = F,
30 header = T,
31 row.names = 1)
32
33 # herein, converting Y matrix to presence-absence
34 Y = 1*(Y>0)
35
36 # select OTUs/species that are present at least in $sp_prevalence (specified above) samples
37 prevalence = colSums(Y != 0)
38 select.sp = prevalence >= sp_prevalence
39 Y = Y[, select.sp]
40 taxonomy = taxonomy[select.sp, ]
41
42 ### creating a phylogenetic tree from a set of nested taxonomic ranks in the taxonomy table
43 # ranks as.factors; assuming that ranks start from the 2nd column in the taxonomy table
44 # and we have 7 ranks
45 for(i in 2:8) taxonomy[,i] = as.factor(taxonomy[,i])
46 # convert tax to phylo tree
47 phy.tree = as.phylo(~Phylum/Class/Order/Family/Genus/Species,
48 data = taxonomy, collapse = FALSE)
49
50 # this "pseudo-tree" does not have any branch lengths, which are needed for the model;
51 # assign arbitrary branch lengths
52 if (is.null(phy.tree$edge.length)) {
53 phy.tree$edge.length = rep(1, nrow(phy.tree$edge))
54 }
55 # rename tree tip lables according to the labels in the Y matrix.
56 phy.tree$tip.label = colnames(Y)
57
58 # check the tree
59 plot(phy.tree, cex=0.6)
Define model
According to our dataset, we are defining the model for HMSC. That is, we specify the structure, including the response variable (community data), covariates (environmental predictors), random effects, and phylogenetic relationships.
1#!/usr/bin/Rscript
2
3library(Hmsc)
4
5 # defining our study design; structure of the data
6 studyDesign = data.frame(
7 sample = as.factor(rownames(X)), # rownames(X) = sample names
8 site = as.factor(X$Site)
9 )
10
11 ### incorporating random effects into the HMSC model.
12 # (to capture the influence of unmeasured factors that vary across different
13 # levels of the data; e.g., among sites, samples).
14 # sampling units
15 rL.sample = HmscRandomLevel(units = levels(studyDesign$sample))
16 # sampling sites
17 rL.site = HmscRandomLevel(units = levels(studyDesign$site))
18
19 # convert sample collection dates into Julian days relative to a specific start date
20 da = as.Date(meta$CollectionDate)
21 jday = 1 + julian(da) - julian(as.Date("2024-01-01"))
22
23 # not covered here, but DOWNLOAD RELEVANT COVARIATES (E.G., CLIMATE, WEATHER, LANDCOVER)
24 # FROM DATABASES BASED ON COORDINATES AND SAMPLING TIMES
25
26 # create a data frame of covariates (predictor variables) that will be included in the model
27 XData = data.frame(seqdepth = log(meta$seq_depth), # number of sequences per sample
28 jday) # Julian days
29
30 ### specify the formula for the fixed effects
31 # using 3.141593 instead of pi to prevent issues when 'pi' is considered as a variable;
32 XFormula = ~cos(2*3.141593*jday/365) + # model seasonal effects; annual cycles
33 sin(2*3.141593*jday/365) + # model seasonal effects; annual cycles
34 cos(4*3.141593*jday/365) + # model seasonal effects; semiannual cycles
35 sin(4*3.141593*jday/365) + # model seasonal effects; semiannual cycles
36 seqdepth # number of sequences per sample
37
38 ### define a model
39 m = Hmsc(Y = Y, # response matrix
40 distr = "probit", # distribution model for the response variable ('probit' for PA)
41 XData = XData, # predictor variables
42 XFormula = XFormula, # fixed effects in the model
43 phyloTree = phy.tree, # phylogenetic tree object
44 studyDesign = studyDesign, # study design object
45 ranLevels = list(sample = rL.sample, site = rL.site)) # random level objects
46
47 # organize, name, and save your HMSC models (to easily manage multiple models if needed)
48 models = list(m)
49 names(models) = c("model_1")
50 save(models, file = paste0("models/unfitted_models.RData"))
51
52 # check models
53 models
Fit model
The following HMSC pipeline is a modified version of the pipeline presented at the ISEC 2024 Hmsc workshop.
input:
Unfitted_models file (unfitted_models.RData)
output:
1#!/usr/bin/Rscript
2
3# set working directory
4localDir = "."
5# specify 'models' dir (input/output dir)
6modelDir = file.path(localDir, "models")
7
8# load input (unfitted_models)
9load(file=file.path(modelDir,"unfitted_models.RData"))
10
11# number of parallel chains (CPUs) for MCMC sampling
12nParallel = NULL # if NULL, then nParallel = nChains
13
14# number of samples and thinning intervals for each iteration
15samples_list = c(5, 250, 250, 250, 250, 250) # number of iterations of the MCMC
16thin_list = c(1, 1, 10, 100, 1000, 10000) # thinning interval; keep every k-th sample
17# number of MCMC chains
18nChains = 4
19#-------------------------------------------------------------------------------------------#
20
21library(Hmsc)
22
23# iterate over sample and thin lists to fit models with different configurations
24nm = length(models) # number of models (for loop)
25if(is.null(nParallel)) nParallel = nChains
26Lst = 1
27while(Lst <= length(samples_list)){
28 thin = thin_list[Lst]
29 samples = samples_list[Lst]
30 print(paste0("thin = ",as.character(thin),"; samples = ", as.character(samples)))
31 filename = file.path(modelDir,paste("models_thin_", as.character(thin),
32 "_samples_", as.character(samples),
33 "_chains_", as.character(nChains),
34 ".Rdata", sep = ""))
35 if(file.exists(filename)){
36 print("model had been fitted already")
37 } else {
38 print(date())
39 for (mi in 1:nm) {
40 print(paste0("model = ",names(models)[mi]))
41 m = models[[mi]]
42 m = sampleMcmc(m, samples = samples, thin=thin,
43 adaptNf=rep(ceiling(0.4*samples*thin), m$nr),
44 transient = ceiling(0.5*samples*thin),
45 nChains = nChains,
46 nParallel = nParallel)
47 models[[mi]] = m
48 }
49 save(models, file=filename)
50 }
51 Lst = Lst + 1
52}
Evaluate convergence
Evaluating convergence ensures the accuracy and reliability of the model’s results by confirming that the MCMC algorithm has adequately explored the parameter space and that the chains have stabilized.
input:
Fitted models directory models.
Check the input path, and typically other parts of the scripts do not need modifications.
output:
MCMC convergence statistics for selected model parameters, illustrated (for all runs) in the file “results/MCMC_convergence.pdf”, and the text file “results/MCMC_convergence.txt”.
1#!/usr/bin/Rscript
2
3library(Hmsc)
4library(colorspace)
5library(vioplot)
6
7# make the script reproducible
8set.seed(1)
9
10# set working directory
11localDir = "."
12# specify 'models' dir (input dir)
13modelDir = file.path(localDir, "models")
14# specify 'results' dir (output dir)
15resultDir = file.path(localDir, "results")
16if (!dir.exists(resultDir)) dir.create(resultDir)
17
18# number of samples and thinning intervals for each iteration (AS ABOVE)
19samples_list = c(5, 250, 250, 250, 250, 250) # number of iterations of the MCMC
20thin_list = c(1, 1, 10, 100, 1000, 10000) # thinning interval; keep every k-th sample
21# number of MCMC chains
22nChains = 4
23#-------------------------------------------------------------------------------------------#
24
25# setting commonly adjusted parameters
26showBeta = TRUE # default = TRUE, converg. shown for beta-parameters
27showGamma = TRUE # default = FALSE, converg. not shown for gamma-parameters
28showOmega = TRUE # default = FALSE, converg. not shown for Omega-parameters
29maxOmega = 100 # default = 50, converg. of Omega shown for 50 randomly selected species pairs
30showRho = TRUE # default = FALSE, converg. not shown for rho-parameters
31showAlpha = TRUE # default = FALSE, converg. not shown for alpha-parameters
32
33# set up a text file to store MCMC convergence statistics
34text.file = file.path(resultDir,"/MCMC_convergence.txt")
35cat("MCMC Convergennce statistics\n\n", file = text.file, sep="")
36
37ma.beta = NULL
38na.beta = NULL
39ma.gamma = NULL
40na.gamma = NULL
41ma.omega= NULL
42na.omega = NULL
43ma.alpha = NULL
44na.alpha = NULL
45ma.rho = NULL
46na.rho = NULL
47Lst = 1
48while(Lst <= nst){
49 thin = thin_list[Lst]
50 samples = samples_list[Lst]
51
52 filename = file.path(modelDir,paste("models_thin_", as.character(thin),
53 "_samples_", as.character(samples),
54 "_chains_",as.character(nChains),
55 ".Rdata",sep = ""))
56 if(file.exists(filename)){
57 load(filename)
58 cat(c("\n",filename,"\n\n"),file=text.file,sep="",append=TRUE)
59 nm = length(models)
60 for(j in 1:nm){
61 mpost = convertToCodaObject(models[[j]], spNamesNumbers = c(T,F), covNamesNumbers = c(T,F))
62 nr = models[[j]]$nr
63 cat(c("\n",names(models)[j],"\n\n"),file=text.file,sep="",append=TRUE)
64 if(showBeta){
65 psrf = gelman.diag(mpost$Beta,multivariate=FALSE)$psrf
66 tmp = summary(psrf)
67 cat("\nbeta\n\n",file=text.file,sep="",append=TRUE)
68 cat(tmp[,1],file=text.file,sep="\n",append=TRUE)
69 if(is.null(ma.beta)){
70 ma.beta = psrf[,1]
71 na.beta = paste0(as.character(thin),",",as.character(samples))
72 } else {
73 ma.beta = cbind(ma.beta,psrf[,1])
74 if(j==1){
75 na.beta = c(na.beta,paste0(as.character(thin),",",as.character(samples)))
76 } else {
77 na.beta = c(na.beta,"")
78 }
79 }
80 }
81 if(showGamma){
82 psrf = gelman.diag(mpost$Gamma,multivariate=FALSE)$psrf
83 tmp = summary(psrf)
84 cat("\ngamma\n\n",file=text.file,sep="",append=TRUE)
85 cat(tmp[,1],file=text.file,sep="\n",append=TRUE)
86 if(is.null(ma.gamma)){
87 ma.gamma = psrf[,1]
88 na.gamma = paste0(as.character(thin),",",as.character(samples))
89 } else {
90 ma.gamma = cbind(ma.gamma,psrf[,1])
91 if(j==1){
92 na.gamma = c(na.gamma,paste0(as.character(thin),",",as.character(samples)))
93 } else {
94 na.gamma = c(na.gamma,"")
95 }
96 }
97 }
98 if(showRho & !is.null(mpost$Rho)){
99 psrf = gelman.diag(mpost$Rho,multivariate=FALSE)$psrf
100 cat("\nrho\n\n",file=text.file,sep="",append=TRUE)
101 cat(psrf[1],file=text.file,sep="\n",append=TRUE)
102 }
103 if(showOmega & nr>0){
104 cat("\nomega\n\n",file=text.file,sep="",append=TRUE)
105 for(k in 1:nr){
106 cat(c("\n",names(models[[j]]$ranLevels)[k],"\n\n"),file=text.file,sep="",append=TRUE)
107 tmp = mpost$Omega[[k]]
108 z = dim(tmp[[1]])[2]
109 if(z > maxOmega){
110 sel = sample(1:z, size = maxOmega)
111 for(i in 1:length(tmp)){
112 tmp[[i]] = tmp[[i]][,sel]
113 }
114 }
115 psrf = gelman.diag(tmp, multivariate = FALSE)$psrf
116 tmp = summary(psrf)
117 cat(tmp[,1],file=text.file,sep="\n",append=TRUE)
118 if(is.null(ma.omega)){
119 ma.omega = psrf[,1]
120 na.omega = paste0(as.character(thin),",",as.character(samples))
121 } else {
122 ma.omega = cbind(ma.omega,psrf[,1])
123 if(j==1){
124 na.omega = c(na.omega,paste0(as.character(thin),",",as.character(samples)))
125 } else {
126 na.omega = c(na.omega,"")
127 }
128 }
129 }
130 }
131 if(showAlpha & nr>0){
132 for(k in 1:nr){
133 if(models[[j]]$ranLevels[[k]]$sDim>0){
134 cat("\nalpha\n\n",file=text.file,sep="\n",append=TRUE)
135 cat(c("\n",names(models[[j]]$ranLevels)[k],"\n\n"),file=text.file,sep="",append=TRUE)
136 psrf = gelman.diag(mpost$Alpha[[k]],multivariate = FALSE)$psrf
137 cat(psrf[,1],file=text.file,sep="\n",append=TRUE)
138 }
139 }
140 }
141 }
142 }
143 Lst = Lst + 1
144}
145
146pdf(file= file.path(resultDir,"/MCMC_convergence.pdf"))
147if(showBeta){
148 par(mfrow=c(2,1))
149 vioplot(ma.beta,col=rainbow_hcl(nm),names=na.beta,ylim=c(0,max(ma.beta)),main="psrf(beta)")
150 legend("topright",legend = names(models), fill=rainbow_hcl(nm))
151 vioplot(ma.beta,col=rainbow_hcl(nm),names=na.beta,ylim=c(0.9,1.1),main="psrf(beta)")
152}
153if(showGamma){
154 par(mfrow=c(2,1))
155 vioplot(ma.gamma,col=rainbow_hcl(nm),names=na.gamma,ylim=c(0,max(ma.gamma)),main="psrf(gamma)")
156 legend("topright",legend = names(models), fill=rainbow_hcl(nm))
157 vioplot(ma.gamma,col=rainbow_hcl(nm),names=na.gamma,ylim=c(0.9,1.1),main="psrf(gamma)")
158}
159if(showOmega & !is.null(ma.omega)){
160 par(mfrow=c(2,1))
161 vioplot(ma.omega,col=rainbow_hcl(nm),names=na.omega,ylim=c(0,max(ma.omega)),main="psrf(omega)")
162 legend("topright",legend = names(models), fill=rainbow_hcl(nm))
163 vioplot(ma.omega,col=rainbow_hcl(nm),names=na.omega,ylim=c(0.9,1.1),main="psrf(omega)")
164}
165dev.off()
check the diagnostics
We need to confirm that the MCMC chain has converged. If the chains have not converged, the samples privided by the MCMC chain can yield biased parameter estimates.
Compute model fit
Assessing the model’s performance and validating its accuracy. …
input:
Fitted models directory models.
Check the input path, and typically other parts of the scripts do not need modifications.
output:
1#!/usr/bin/Rscript
2
3library(Hmsc)
4
5# make the script reproducible
6set.seed(1)
7
8# set working directory
9localDir = "."
10# specify 'models' dir (input/output dir)
11modelDir = file.path(localDir, "models")
12
13# number of samples and thinning intervals for each iteration (AS ABOVE)
14samples_list = c(5, 250, 250, 250, 250, 250) # number of iterations of the MCMC
15thin_list = c(1, 1, 10, 100, 1000, 10000) # thinning interval; keep every k-th sample
16# number of MCMC chains
17nChains = 4
18
19# number of parallel chains (CPUs) for MCMC sampling
20nParallel = NULL # if NULL, then nParallel = nChains
21# number of cross-validations
22nfolds = 2 # default: (2) two-fold cross-validation
23#-------------------------------------------------------------------------------------------#
24
25if(is.null(nParallel)) nParallel = nChains
26Lst = 1
27while(Lst <= length(samples_list)){
28 thin = thin_list[Lst]
29 samples = samples_list[Lst]
30 filename.in = file.path(modelDir,paste("models_thin_", as.character(thin),
31 "_samples_", as.character(samples),
32 "_chains_",as.character(nChains),
33 ".Rdata",sep = ""))
34 filename.out = file.path(modelDir,paste("MF_thin_", as.character(thin),
35 "_samples_", as.character(samples),
36 "_chains_",as.character(nChains),
37 "_nfolds_", as.character(nfolds),
38 ".Rdata",sep = ""))
39 if(file.exists(filename.out)){
40 print(paste0("thin = ",as.character(thin),"; samples = ",as.character(samples)))
41 print("model fit had been computed already")
42 } else {
43 if(file.exists(filename.in)){
44 print(paste0("thin = ",as.character(thin),"; samples = ",as.character(samples)))
45 print(date())
46 load(file = filename.in) #models
47 nm = length(models)
48
49 MF = list()
50 MFCV = list()
51 WAIC = list()
52
53 for(mi in 1:nm){
54 print(paste0("model = ",names(models)[mi]))
55 m = models[[mi]]
56 preds = computePredictedValues(m)
57 MF[[mi]] = evaluateModelFit(hM=m, predY=preds)
58 partition = createPartition(m, nfolds = nfolds) #USE column = ...
59 preds = computePredictedValues(m,partition=partition, nParallel = nParallel)
60 MFCV[[mi]] = evaluateModelFit(hM=m, predY=preds)
61 WAIC[[mi]] = computeWAIC(m)
62 }
63 names(MF)=names(models)
64 names(MFCV)=names(models)
65 names(WAIC)=names(models)
66
67 save(MF,MFCV,WAIC,file = filename.out)
68 }
69 }
70 Lst = Lst + 1
71}
Show model fit
Evaluate and visualize the model fit for different thinning and sampling configurations.
input:
Fitted models directory models.
Check the input path, and typically other parts of the scripts do not need modifications.
output:
Model fits illustrated (for highest RUN of compute model fit) in the file results/model_fit.pdf.
1#!/usr/bin/Rscript
2
3library(Hmsc)
4
5# make the script reproducible
6set.seed(1)
7
8# set working directory
9localDir = "."
10# specify 'models' dir (input dir)
11modelDir = file.path(localDir, "models")
12# specify 'results' dir (output dir)
13resultDir = file.path(localDir, "results")
14if (!dir.exists(resultDir)) dir.create(resultDir)
15
16# number of samples and thinning intervals for each iteration (AS ABOVE)
17samples_list = c(5, 250, 250, 250, 250, 250) # number of iterations of the MCMC
18thin_list = c(1, 1, 10, 100, 1000, 10000) # thinning interval; keep every k-th sample
19# number of MCMC chains
20nChains = 4
21
22# number of cross-validations
23nfolds = 2 # default: (2) two-fold cross-validation
24#-------------------------------------------------------------------------------------------#
25
26for (Lst in nst:1) {
27 thin = thin_list[Lst]
28 samples = samples_list[Lst]
29
30 filename = file.path(modelDir,paste("MF_thin_", as.character(thin),
31 "_samples_", as.character(samples),
32 "_chains_",as.character(nChains),
33 "_nfolds_", as.character(nfolds),
34 ".Rdata",sep = ""))
35 if(file.exists(filename)){break}
36}
37if(file.exists(filename)){
38 load(filename)
39
40 nm = length(MF)
41 modelnames = names(MF)
42 pdf(file = file.path(resultDir,paste0("/model_fit_nfolds_",nfolds,".pdf")))
43 for(j in 1:nm){
44 cMF = MF[[j]]
45 cMFCV = MFCV[[j]]
46 if(!is.null(cMF$TjurR2)){
47 plot(cMF$TjurR2,cMFCV$TjurR2,xlim=c(-1,1),ylim=c(-1,1),
48 xlab = "explanatory power",
49 ylab = "predictive power",
50 main=paste0(modelnames[j],", thin = ",
51 as.character(thin),
52 ", samples = ",as.character(samples),
53 ": Tjur R2.\n",
54 "mean(MF) = ",as.character(mean(cMF$TjurR2,na.rm=TRUE)),
55 ", mean(MFCV) = ",as.character(mean(cMFCV$TjurR2,na.rm=TRUE))))
56 abline(0,1)
57 abline(v=0)
58 abline(h=0)
59 }
60 if(!is.null(cMF$R2)){
61 plot(cMF$R2,cMFCV$R2,xlim=c(-1,1),ylim=c(-1,1),
62 xlab = "explanatory power",
63 ylab = "predictive power",
64 main=paste0(modelnames[[j]],", thin = ",as.character(thin),
65 ", samples = ",as.character(samples),
66 ": R2. \n",
67 "mean(MF) = ",as.character(mean(cMF$R2,na.rm=TRUE)),
68 ", mean(MFCV) = ",as.character(mean(cMFCV$R2,na.rm=TRUE))))
69 abline(0,1)
70 abline(v=0)
71 abline(h=0)
72 }
73 if(!is.null(cMF$AUC)){
74 plot(cMF$AUC,cMFCV$AUC,xlim=c(0,1),ylim=c(0,1),
75 xlab = "explanatory power",
76 ylab = "predictive power",
77 main=paste0(modelnames[[j]],", thin = ",as.character(thin),
78 ", samples = ",as.character(samples),
79 ": AUC. \n",
80 "mean(MF) = ",as.character(mean(cMF$AUC,na.rm=TRUE)),
81 ", mean(MFCV) = ",as.character(mean(cMFCV$AUC,na.rm=TRUE))))
82 abline(0,1)
83 abline(v=0.5)
84 abline(h=0.5)
85 }
86 if(FALSE && !is.null(cMF$O.TjurR2)){
87 plot(cMF$O.TjurR2,cMFCV$O.TjurR2,xlim=c(-1,1),ylim=c(-1,1),
88 xlab = "explanatory power",
89 ylab = "predictive power",
90 main=paste0(modelnames[[j]],", thin = ",as.character(thin),", samples = ",as.character(samples),": O.Tjur R2"))
91 abline(0,1)
92 abline(v=0)
93 abline(h=0)
94 }
95 if(FALSE && !is.null(cMF$O.AUC)){
96 plot(cMF$O.AUC,cMFCV$O.AUC,xlim=c(0,1),ylim=c(0,1),
97 xlab = "explanatory power",
98 ylab = "predictive power",
99 main=paste0(modelnames[[j]],", thin = ",as.character(thin),", samples = ",as.character(samples),": O.AUC"))
100 abline(0,1)
101 abline(v=0.5)
102 abline(h=0.5)
103 }
104 if(!is.null(cMF$SR2)){
105 plot(cMF$SR2,cMFCV$SR2,xlim=c(-1,1),ylim=c(-1,1),
106 xlab = "explanatory power",
107 ylab = "predictive power",
108 main=paste0(modelnames[[j]],", thin = ",as.character(thin),
109 ", samples = ",as.character(samples),
110 ": SR2. \n",
111 "mean(MF) = ",as.character(mean(cMF$SR2,na.rm=TRUE)),
112 ", mean(MFCV) = ",as.character(mean(cMFCV$SR2,na.rm=TRUE))))
113 abline(0,1)
114 abline(v=0)
115 abline(h=0)
116 }
117 if(FALSE && !is.null(cMF$C.SR2)){
118 plot(cMF$C.SR2,cMFCV$C.SR2,xlim=c(-1,1),ylim=c(-1,1),
119 xlab = "explanatory power",
120 ylab = "predictive power",
121 main=paste0(modelnames[[j]],", thin = ",as.character(thin),", samples = ",as.character(samples),": C.SR2"))
122 abline(0,1)
123 abline(v=0)
124 abline(h=0)
125 }
126 }
127 dev.off()
128}
Show parameter estimates
Assessing the model’s performance and validating its accuracy. …
input:
Fitted models directory models.
Check the input path, and typically other parts of the scripts do not need modifications.
output:
1#!/usr/bin/Rscript
2
3library(Hmsc)
4library(colorspace)
5library(corrplot)
6library(writexl)
7
8# make the script reproducible
9set.seed(1)
10
11# set working directory
12localDir = "."
13# specify 'models' dir (input dir)
14modelDir = file.path(localDir, "models")
15# specify 'results' dir (output dir)
16resultDir = file.path(localDir, "results")
17if (!dir.exists(resultDir)) dir.create(resultDir)
18
19# number of samples and thinning intervals for each iteration (AS ABOVE)
20samples_list = c(5, 250, 250, 250, 250, 250) # number of iterations of the MCMC
21thin_list = c(1, 1, 10, 100, 1000, 10000) # thinning interval; keep every k-th sample
22# number of MCMC chains
23nChains = 4
24
25# thresholds for the posterior support levels
26support.level.beta = 0.95 # default: 0.95
27support.level.gamma = 0.95 # default: 0.95
28support.level.omega = 0.9 # default: 0.9
29
30
31var.part.order.explained = NULL # default: in variance partitioning of explained variance,
32 # species are shown in the order they are in the model.
33 # -> use var.part.order.explained to select which order species are shown in the
34 # raw variance partitioning.
35 # var.part.order.raw should be a list of length the number of models.
36 # for each element provide either 0 (use default);
37 # or a vector of species indices;
38 # or "decreasing" if you wish to order according to explanatory power
39 # var.part.order.explained = list()
40 # var.part.order.explained[[1]] = 0
41 # var.part.order.explained[[2]] = c(2,1)
42
43var.part.order.raw = NULL # default: in variance partitioning of raw variance, species
44 # are shown in the order they are in the model.
45 # -> use var.part.order.raw to select which order species are shown in the
46 # explained variance partitioning.
47 # same options apply as for var.part.order.explained
48 # var.part.order.raw = list()
49 # var.part.order.raw[[1]] = "decreasing"
50 # var.part.order.raw[[2]] = c(1,2)
51
52
53show.sp.names.beta = NULL # default: species names shown in beta plot if there are at
54 # most 30 species and no phylogeny
55 # -> use show.sp.names.beta to choose to show / not show species names in betaPlot
56 # if given, show.sp.names.beta should be a vector with length equalling number of models
57 # show.sp.names.beta = c(TRUE,FALSE)
58
59plotTree = NULL # default: tree is plotted in Beta plot if the model includes it.
60 # -> use plotTree to choose to plot / not plot the tree in betaPlot.
61 # if given, plotTree should be a vector with length equalling number of models
62 # plotTree = c(FALSE,FALSE)
63
64omega.order = NULL # default: species shown in the order they are in the model.
65 # -> use omega.order to select which order species are shown in omega plots
66 # omega.order should be a list of length the number of models.
67 # for each element provide either 0 (use default);
68 # or a vector of species indices;
69 # or "AOE" if you wish to use the angular order of the eigenvectors.
70 # omega.order = list()
71 # omega.order[[1]] = "AOE"
72 # omega.order[[2]] = c(2,1)
73
74show.sp.names.omega = NULL # default: species names shown in beta plot if there are at
75 # most 30 species.
76 # show.sp.names.omega = c(TRUE,FALSE)
77#-------------------------------------------------------------------------------------------#
78
79
80text.file = file.path(resultDir,"/parameter_estimates.txt")
81cat(c("This file contains additional information regarding parameter estimates.","\n","\n",
82 sep=""), file=text.file)
83
84for (Lst in nst:1) {
85 thin = thin_list[Lst]
86 samples = samples_list[Lst]
87 filename = file.path(modelDir,paste("models_thin_", as.character(thin),
88 "_samples_", as.character(samples),
89 "_chains_",as.character(nChains),
90 ".Rdata",sep = ""))
91 if(file.exists(filename)){break}
92}
93if(file.exists(filename)){
94 load(filename)
95 cat(c("\n",filename,"\n","\n"),file=text.file,sep="",append=TRUE)
96 nm = length(models)
97 if(is.null(var.part.order.explained)){
98 var.part.order.explained = list()
99 for(j in 1:nm) var.part.order.explained[[j]] = 0
100 }
101 if(is.null(var.part.order.raw)){
102 var.part.order.raw = list()
103 for(j in 1:nm) var.part.order.raw[[j]] = 0
104 }
105 if(is.null(omega.order)){
106 omega.order = list()
107 for(j in 1:nm) omega.order[[j]] = 0
108 }
109
110 modelnames = names(models)
111
112 pdf(file= file.path(resultDir,"parameter_estimates.pdf"))
113 for(j in 1:nm){
114 cat(c("\n",names(models)[j],"\n","\n"),file=text.file,sep="",append=TRUE)
115 m = models[[j]]
116 if(m$XFormula=="~."){
117 covariates = colnames(m$X)[-1]
118 } else {
119 covariates = attr(terms(m$XFormula),"term.labels")
120 }
121 if(m$nr+length(covariates)>1 & m$ns>1){
122 preds = computePredictedValues(m)
123 VP = computeVariancePartitioning(m)
124 vals = VP$vals
125 mycols = rainbow(nrow(VP$vals))
126 MF = evaluateModelFit(hM=m, predY=preds)
127 R2 = NULL
128 if(!is.null(MF$TjurR2)){
129 TjurR2 = MF$TjurR2
130 vals = rbind(vals,TjurR2)
131 R2=TjurR2
132 }
133 if(!is.null(MF$R2)){
134 R2=MF$R2
135 vals = rbind(vals,R2)
136 }
137 if(!is.null(MF$SR2)){
138 R2=MF$SR2
139 vals = rbind(vals,R2)
140 }
141 filename = file.path(resultDir, paste("parameter_estimates_VP_",modelnames[j],".csv"))
142 write.csv(vals,file=filename)
143 if(!is.null(VP$R2T$Beta)){
144 filename = file.path(resultDir,paste("parameter_estimates_VP_R2T_Beta",modelnames[j],
145 ".csv"))
146 write.csv(VP$R2T$Beta,file=filename)
147 }
148 if(!is.null(VP$R2T$Y)){
149 filename = file.path(resultDir, paste("parameter_estimates_VP_R2T_Y",modelnames[j],
150 ".csv"))
151 write.csv(VP$R2T$Y,file=filename)
152 }
153 if(all(var.part.order.explained[[j]]==0)){
154 c.var.part.order.explained = 1:m$ns
155 } else {
156 if(all(var.part.order.explained[[j]]=="decreasing")){
157 c.var.part.order.explained = order(R2, decreasing = TRUE)
158 }
159 else {
160 c.var.part.order.explained = var.part.order.explained[[j]]
161 }
162 }
163 VP$vals = VP$vals[,c.var.part.order.explained]
164 cat(c("\n","var.part.order.explained","\n","\n"),file=text.file,sep="",append=TRUE)
165 cat(m$spNames[c.var.part.order.explained],file=text.file,sep="\n",append=TRUE)
166 plotVariancePartitioning(hM=m, VP=VP, main = paste0("Proportion of explained variance, ",
167 modelnames[j]), cex.main=0.8, cols = mycols,
168 args.leg=list(bg="white",cex=0.7))
169 if(all(var.part.order.raw[[j]]==0)){
170 c.var.part.order.raw = 1:m$ns
171 } else {
172 if(all(var.part.order.raw[[j]]=="decreasing")){
173 c.var.part.order.raw = order(R2, decreasing = TRUE)
174 }
175 else {
176 c.var.part.order.raw = var.part.order.raw[[j]]
177 }
178 }
179 if(!is.null(R2)){
180 VPr = VP
181 for(k in 1:m$ns){
182 VPr$vals[,k] = R2[k]*VPr$vals[,k]
183 }
184 VPr$vals = VPr$vals[,c.var.part.order.raw]
185 cat(c("\n","var.part.order.raw","\n","\n"),file=text.file,sep="",append=TRUE)
186 cat(m$spNames[c.var.part.order.raw],file=text.file,sep="\n",append=TRUE)
187 plotVariancePartitioning(hM=m, VP=VPr,main=paste0("Proportion of raw variance, ",
188 modelnames[j]),cex.main=0.8, cols = mycols, args.leg=list(bg="white",cex=0.7),
189 ylim=c(0,1))
190 }
191 }
192 }
193 for(j in 1:nm){
194 m = models[[j]]
195 if(m$nc>1){
196 postBeta = getPostEstimate(m, parName="Beta")
197 filename = file.path(resultDir,
198 paste("parameter_estimates_Beta_",modelnames[j],".xlsx"))
199 me = as.data.frame(t(postBeta$mean))
200 me = cbind(m$spNames,me)
201 colnames(me) = c("Species",m$covNames)
202 po = as.data.frame(t(postBeta$support))
203 po = cbind(m$spNames,po)
204 colnames(po) = c("Species",m$covNames)
205 ne = as.data.frame(t(postBeta$supportNeg))
206 ne = cbind(m$spNames,ne)
207 colnames(ne) = c("Species",m$covNames)
208 vals = list("Posterior mean"=me,"Pr(x>0)"=po,"Pr(x<0)"=ne)
209 writexl::write_xlsx(vals,path = filename)
210 if(is.null(show.sp.names.beta)){
211 c.show.sp.names = (is.null(m$phyloTree) && m$ns<=30)
212 } else {
213 c.show.sp.names = show.sp.names.beta[j]
214 }
215 c.plotTree = !is.null(m$phyloTree)
216 if(!is.null(plotTree)){
217 c.plotTree = c.plotTree & plotTree[j]
218 }
219 plotBeta(m, post=postBeta, supportLevel = support.level.beta, param="Sign",
220 plotTree = c.plotTree,
221 covNamesNumbers = c(TRUE,FALSE),
222 spNamesNumbers=c(c.show.sp.names,FALSE),
223 cex=c(0.6,0.6,0.8))
224 mymain = paste0("BetaPlot, ",modelnames[j])
225 if(!is.null(m$phyloTree)){
226 mpost = convertToCodaObject(m)
227 rhovals = unlist(poolMcmcChains(mpost$Rho))
228 mymain = paste0(mymain,", E[rho] = ",round(mean(rhovals),2),", Pr[rho>0] = ",
229 round(mean(rhovals>0),2))
230 }
231 title(main=mymain, line=2.5, cex.main=0.8)
232 }
233 }
234 for(j in 1:nm){
235 m = models[[j]]
236 if(m$nt>1 & m$nc>1){
237 postGamma = getPostEstimate(m, parName="Gamma")
238 plotGamma(m, post=postGamma, supportLevel = support.level.gamma, param="Sign",
239 covNamesNumbers = c(TRUE,FALSE),
240 trNamesNumbers=c(m$nt<21,FALSE),
241 cex=c(0.6,0.6,0.8))
242 title(main=paste0("GammaPlot ",modelnames[j]), line=2.5,cex.main=0.8)
243 }
244 }
245 for(j in 1:nm){
246 m = models[[j]]
247 if(m$nr>0 & m$ns>1){
248 OmegaCor = computeAssociations(m)
249 for (r in 1:m$nr){
250 toPlot = ((OmegaCor[[r]]$support>support.level.omega) +
251 (OmegaCor[[r]]$support<(1-support.level.omega))>0)*sign(OmegaCor[[r]]$mean)
252 if(is.null(show.sp.names.omega)){
253 c.show.sp.names = (m$ns<=30)
254 } else {
255 c.show.sp.names = show.sp.names.omega[j]
256 }
257 if(!c.show.sp.names){
258 colnames(toPlot)=rep("",m$ns)
259 rownames(toPlot)=rep("",m$ns)
260 }
261 if(all(omega.order[[j]]==0)){
262 plotOrder = 1:m$ns
263 } else {
264 if(all(omega.order[[j]]=="AOE")){
265 plotOrder = corrMatOrder(OmegaCor[[r]]$mean,order="AOE")
266 } else {
267 plotOrder = omega.order[[j]]
268 }
269 }
270 cat(c("\n","omega.order","\n","\n"),file=text.file,sep="",append=TRUE)
271 cat(m$spNames[plotOrder],file=text.file,sep="\n",append=TRUE)
272 mymain = paste0("Associations, ",modelnames[j], ": ",names(m$ranLevels)[[r]])
273 if(m$ranLevels[[r]]$sDim>0){
274 mpost = convertToCodaObject(m)
275 alphavals = unlist(poolMcmcChains(mpost$Alpha[[1]][,1]))
276 mymain = paste0(mymain,", E[alpha1] = ",round(mean(alphavals),2),", Pr[alpha1>0] = ",
277 round(mean(alphavals>0),2))
278 }
279 corrplot(toPlot[plotOrder,plotOrder], method = "color",
280 col=colorRampPalette(c("blue","white","red"))(3),
281 mar=c(0,0,1,0),
282 main=mymain,cex.main=0.8)
283 me = as.data.frame(OmegaCor[[r]]$mean)
284 me = cbind(m$spNames,me)
285 colnames(me)[1] = ""
286 po = as.data.frame(OmegaCor[[r]]$support)
287 po = cbind(m$spNames,po)
288 colnames(po)[1] = ""
289 ne = as.data.frame(1-OmegaCor[[r]]$support)
290 ne = cbind(m$spNames,ne)
291 colnames(ne)[1] = ""
292 vals = list("Posterior mean"=me,"Pr(x>0)"=po,"Pr(x<0)"=ne)
293 filename = file.path(resultDir, paste("parameter_estimates_Omega_",modelnames[j],"_",
294 names(m$ranLevels)[[r]],".xlsx"))
295 writexl::write_xlsx(vals,path = filename)
296 }
297 }
298 }
299 dev.off()
300}
Make predictions
Making predictions and generating plots.
input:
Fitted models directory models.
Check the input path, and typically other parts of the scripts do not need modifications.
output:
Predictions over environmental gradients (for highest RUN of Fit models) in the file results/predictions.pdf.
1#!/usr/bin/Rscript
2
3library(Hmsc)
4library(ggplot2)
5
6# make the script reproducible
7set.seed(1)
8
9# set working directory
10localDir = "."
11# specify 'models' dir (input dir)
12modelDir = file.path(localDir, "models")
13# specify 'results' dir (output dir)
14resultDir = file.path(localDir, "results")
15if (!dir.exists(resultDir)) dir.create(resultDir)
16
17# number of samples and thinning intervals for each iteration (AS ABOVE)
18samples_list = c(5, 250, 250, 250, 250, 250) # number of iterations of the MCMC
19thin_list = c(1, 1, 10, 100, 1000, 10000) # thinning interval; keep every k-th sample
20# number of MCMC chains
21nChains = 4
22
23species.list = NULL # one example species shown for each model,
24# selected as prevalence closest to 0.5 (probit models)
25# or most abundant species (other models).
26 # -> use species.list to select which species are used as examples for which
27 # predictions are shown
28 # species.list should be a list of length the number of models.
29 # for each element provide either 0 (use default) or a vector of species indices
30 # species.list = list()
31 # species.list[[1]] = 0
32 # species.list[[2]] = c(1,2)
33
34trait.list = NULL # community weighted mean shown for all traits.
35 # -> use trait.list to select for which traits predictions for community weighted
36 # mean traits are shown
37 # trait.list should be a list of length the number of models.
38 # for each element provide either 0 (use default) or a vector of trait indices
39 # see models[[j]]$trNames to see which trait each index corresponds to
40 # trait.list = list()
41 # trait.list[[1]] = c(2,10)
42 # trait.list[[2]] = 0
43
44env.list = NULL # predictions constructed over all environmental gradients.
45 # -> use env.list to select over which environmental gradients predictions are generated
46 # env.list should be a list of length the number of models.
47 # for each element provide either 0 (use default) or a vector of environmental variables
48 # env.list = list()
49 # env.list[[1]] = 0
50 # env.list[[2]] = c("tree","decay")
51#-------------------------------------------------------------------------------------------#
52
53for (Lst in nst:1) {
54 thin = thin_list[Lst]
55 samples = samples_list[Lst]
56 filename = file.path(modelDir,paste("models_thin_", as.character(thin),
57 "_samples_", as.character(samples),
58 "_chains_",as.character(nChains),
59 ".Rdata",sep = ""))
60 if(file.exists(filename)){break}
61}
62if(file.exists(filename)){
63 load(filename)
64 nm = length(models)
65 modelnames = names(models)
66 if(is.null(species.list)){
67 species.list = list()
68 for(j in 1:nm) species.list[[j]] = 0
69 }
70 if(is.null(trait.list)){
71 trait.list = list()
72 for(j in 1:nm) trait.list[[j]] = 0
73 }
74 if(is.null(env.list)){
75 env.list = list()
76 for(j in 1:nm) env.list[[j]] = 0
77 }
78
79 pdf(file= file.path(resultDir,"predictions.pdf"))
80 for(j in 1:nm){
81 m = models[[j]]
82 if(all(env.list[[j]]==0)){
83 if(m$XFormula=="~."){
84 covariates = colnames(m$XData)
85 } else {
86 covariates = all.vars(m$XFormula)
87 }
88 } else {
89 covariates = env.list[[j]]
90 }
91 covariates = setdiff(covariates,"pi")
92 ex.sp = which.max(colMeans(m$Y,na.rm = TRUE)) #most common species as example species
93 if(m$distr[1,1]==2){
94 ex.sp = which.min(abs(colMeans(m$Y,na.rm = TRUE)-0.5))
95 }
96 if(!all(species.list[[j]])==0){
97 ex.sp = species.list[[j]]
98 }
99 if(length(covariates)>0){
100 for(k in 1:(length(covariates))){
101 covariate = covariates[[k]]
102 Gradient = constructGradient(m,focalVariable = covariate,ngrid = 100)
103 Gradient2 = constructGradient(m,focalVariable = covariate,non.focalVariables = 1,
104 ngrid = 100)
105 predY = predict(m, Gradient=Gradient, expected = TRUE)
106 predY2 = predict(m, Gradient=Gradient2, expected = TRUE)
107 par(mfrow=c(2,1))
108 pl = plotGradient(m, Gradient, pred=predY, yshow = 0, measure="S", showData = TRUE,
109 main = paste0(modelnames[j],": summed response (total effect)"))
110 if(inherits(pl, "ggplot")){
111 print(pl + labs(title=paste0(modelnames[j],": summed response (total effect)")))
112 }
113 pl = plotGradient(m, Gradient2, pred=predY2, yshow = 0, measure="S", showData = TRUE,
114 main = paste0(modelnames[j],": summed response (marginal effect)"))
115 if(inherits(pl, "ggplot")){
116 print(pl + labs(title=paste0(modelnames[j],": summed response (marginal effect)")))
117 }
118 for(l in 1:length(ex.sp)){
119 par(mfrow=c(2,1))
120 pl = plotGradient(m, Gradient, pred=predY,
121 yshow = if(m$distr[1,1]==2){c(-0.1,1.1)}else{0},
122 measure="Y",index=ex.sp[l],
123 showData = TRUE,
124 main = paste0(modelnames[j],": example species (total effect)"))
125 if(inherits(pl, "ggplot")){
126 print(pl + labs(title=paste0(modelnames[j],": example species (total effect)")))
127 }
128 pl = plotGradient(m, Gradient2, pred=predY2,
129 yshow = if(m$distr[1,1]==2){c(-0.1,1.1)}else{0},
130 measure="Y",index=ex.sp[l], showData = TRUE,
131 main = paste0(modelnames[j],": example species (marginal effect)"))
132 if(inherits(pl, "ggplot")){
133 print(pl + labs(title=paste0(modelnames[j],": example species (marginal effect)")))
134 }
135 }
136 if(m$nt>1){
137 traitSelection = 2:m$nt
138 if(!all(trait.list[[j]]==0)) traitSelection = trait.list[[j]]
139 for(l in traitSelection){
140 par(mfrow=c(2,1))
141 pl = plotGradient(m, Gradient, pred=predY,
142 measure="T", index=l, showData = TRUE,yshow = 0,
143 main = paste0(modelnames[j],": community weighted mean trait
144 (total effect)"))
145 if(inherits(pl, "ggplot")){
146 print(pl + labs(title=paste0(modelnames[j],": community weighted mean trait
147 (total effect)")))
148 }
149 pl = plotGradient(m, Gradient2, pred=predY2,
150 measure="T", index=l, showData = TRUE, yshow = 0,
151 main = paste0(modelnames[j],": community weighted mean trait
152 (marginal effect)"))
153 if(inherits(pl, "ggplot")){
154 print(pl + labs(title=paste0(modelnames[j],": community weighted mean trait
155 (marginal effect)")))
156 }
157 }
158 }
159 }
160 }
161 }
162 dev.off()
163}
![]()

![]()
![]()