![]()
Fungi/ITS
This is executable step-by-step pipeline for ITS2 amplicon data from Illumina sequencing machine. The bioinformatic workflow results in amplicon sequence variants (ASVs) and well as operational taxonomic units (OTUs).
Note
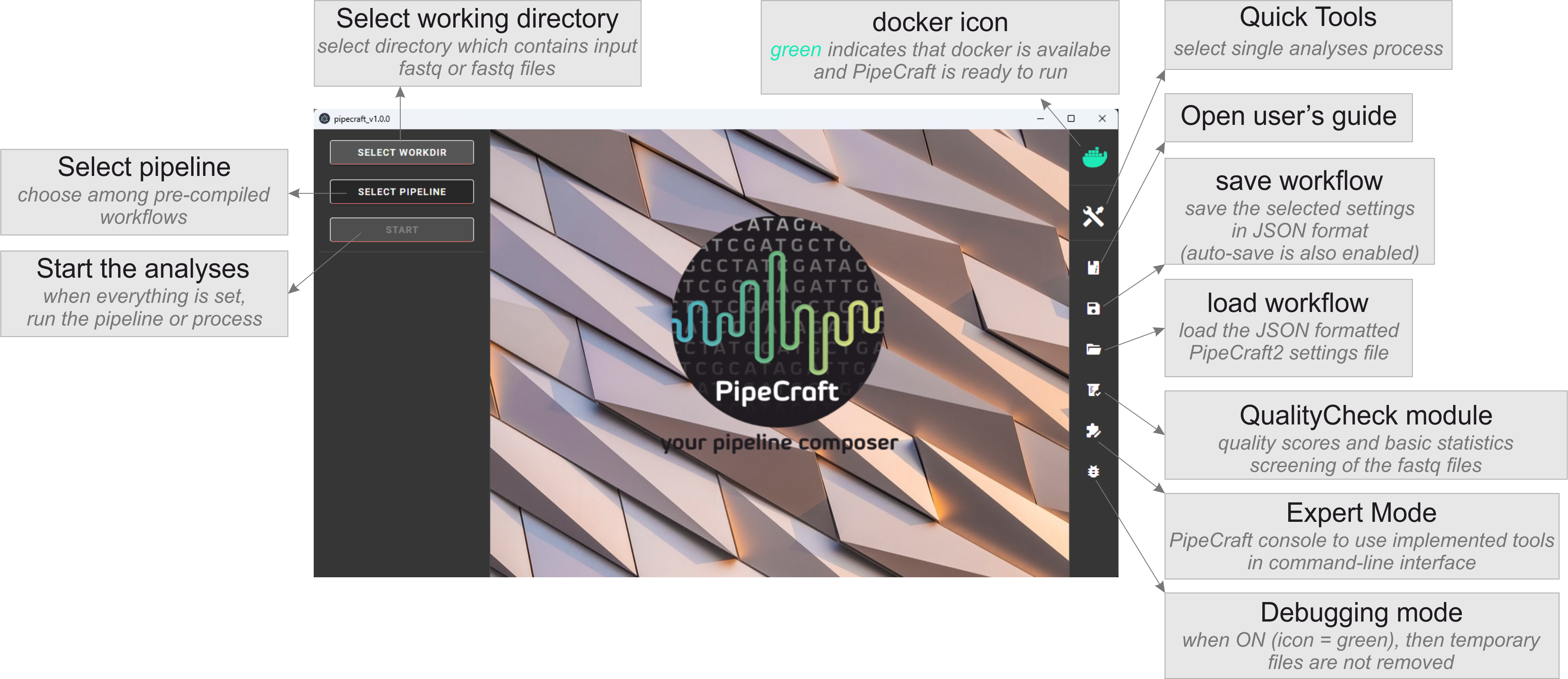
The full bioinformatics workflow can be automatically run through PipeCraft2, a software package which represents a graphical user interface wrapper for all the bioinformatic steps below. See the example workflow for ITS2.
Citation of the pipeline
When using this pipeline, please cite as: Anslan, S, Bahram, M, Hiiesalu, I, Tedersoo, L. PipeCraft: Flexible open-source toolkit for bioinformatics analysis of custom high-throughput amplicon sequencing data. Mol Ecol Resour. 2017; 17: e234-e240. https://doi.org/10.1111/1755-0998.12692
Also, please cite the original resources of the wrapped software.
Dependencies
Process |
Software |
Version |
|---|---|---|
cutadapt |
4.4 |
|
DADA2 |
2.28 |
|
DADA2 |
2.28 |
|
DADA2 |
2.28 |
|
DADA2 |
2.28 |
|
UNCROSS2 |
||
ITSx |
1.1.3 |
|
vsearch |
2.28.1 |
|
BLAST |
2.12.0+ |
|
LULU |
0.1.0+ |
*only applicable when there are multiple sequencing runs per study.
Note
All the dependencies/software of the pipeline are available on a Docker image.
docker pull pipecraft/bioscanflow:2
# run docker
# specify the files location with -v flag ($PWD = the current working directory)
docker run -i --tty -v $PWD/:/Files pipecraft/bioscanflow:2
# inside the container, the files are accessible in the /Files dir
cd Files
# checking if cutadapt is available
cutadapt -h
# ready to run the pipe as below ...
## make sure that via the shared folder (-v) path you have access also to the reference databases.
Data structure
Multiple sequencing runs
Important
When aiming to combine samples from multiple sequencing runs, then follow the below directory structure
Directory structure:
Note
Fastq files with the same name will be considered as the same sample and will be merged in the “Merge sequencing runs” step.
Single sequencing run
Remove primers
Note
Here, assuming that all sequences are in 5’-3’ orientation! (3’-5’ orient sequences will be discarded with this workflow)
Important
1 #!/bin/bash
2 ## workflow to remove primers via cutadapt
3
4 # My working folder = /multiRunDir (see dir structure above)
5
6 # specify the identifier string for the R1 files
7 read_R1="_R1"
8
9 ## specify primers
10 # ITS2 primers
11 fwd_primer=$"GTGARTCATCRARTYTTTG" #this is primer gITS7ngs
12 rev_primer=$"CCTSCSCTTANTDATATGC" #this is primer ITS4ngsUni
13 # ITS1 primers
14 #fwd_primer=$"CTTGGTCATTTAGAGGAAGTAA" #this is primer ITS1F
15 #rev_primer=$"GCTGCGTTCTTCATCGATGC" #this is primer ITS2
16
17
18
19 # edit primer trimming settings
20 maximum_error_rate="2" # Maximum error rate in primer string search;
21 # if set as 1, then allow 1 mismatch;
22 # if set as 0.1, then allow mismatch in 10% of the bases,
23 # i.e. if a primer is 20 bp then allowing 2 mismatches.
24 overlap="17" # The minimum overlap length. Keep it nearly as high
25 # as the primer length to avoid short random matches.
26 pair_filter="both" # Option 'any' discards a read pair if primers are not found in
27 # either of the read pairs (R1 and R2).
28 # Option 'both' keeps the read pair if a primer is found in
29 # at least one of the read pairs.
30
31 # get directory names if working with multiple sequencing runs
32 DIRS=$(ls -d *) # -> sequencing_set01 sequencing_set02 sequencing_set03
33
34 for sequencing_run in $DIRS; do
35 printf "\nWorking with $sequencing_run \n"
36 cd $sequencing_run
37 #-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#
38 # make output dirs
39 mkdir -p primersCut_out
40 mkdir -p primersCut_out/untrimmed
41
42 ### Clip primers with cutadapt
43 for inputR1 in *$read_R1*; do
44 inputR2=$(echo $inputR1 | sed -e 's/R1/R2/')
45 cutadapt --quiet \
46 -e $maximum_error_rate \
47 --minimum-length 32 \
48 --overlap $overlap \
49 --no-indels \
50 --cores=0 \
51 --untrimmed-output primersCut_out/untrimmed/$inputR1 \
52 --untrimmed-paired-output primersCut_out/untrimmed/$inputR2 \
53 --pair-filter=$pair_filter \
54 -g $fwd_primer \
55 -G $rev_primer \
56 -o primersCut_out/$inputR1 \
57 -p primersCut_out/$inputR2 \
58 $inputR1 $inputR2
59 done
60 #-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#
61 cd ..
62 done
Quality filtering
1 #!/usr/bin/Rscript
2 ## workflow to perform quality filtering within DADA2
3
4 #load dada2 library
5 library('dada2')
6
7 # specify the identifier string for the R1 files
8 read_R1 = "_R1"
9
10 # get the identifier string for the R2 files
11 read_R2 = gsub("R1", "R2", read_R1)
12
13 # capturing the directory structure when working with multiple runs
14 wd = getwd() # -> wd is "~/multiRunDir"
15 dirs = list.dirs(recursive = FALSE)
16 for (i in 1:length(dirs)) {
17 if(length(dirs) > 1) {
18 setwd(dirs[i])
19 print(paste0("Working with ", dirs[i]))
20 #-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#
21 # output path
22 path_results = "qualFiltered_out"
23 # input and output file paths
24 R1s = sort(list.files("primersCut_out", pattern = read_R1, full.names = TRUE))
25 R2s = sort(list.files("primersCut_out", pattern = read_R2, full.names = TRUE))
26 #sample names
27 sample_names = sapply(strsplit(basename(R1s), read_R1), `[`, 1)
28
29 # filtered files path
30 filtR1 = file.path(path_results, paste0(sample_names, ".R1.", "fastq.gz"))
31 filtR2 = file.path(path_results, paste0(sample_names, ".R2.", "fastq.gz"))
32 names(filtR1) = sample_names
33 names(filtR2) = sample_names
34
35 #quality filtering
36 qfilt = filterAndTrim(R1s, filtR1, R2s, filtR2,
37 maxN = 0, # max number of allowed N bases.
38 maxEE = c(2, 2), # max error rate per R1 and R2 read, respectively.
39 truncQ = 2, # truncate reads at the first instance of a quality score less than or equal to specified value.
40 truncLen = c(0, 0), # truncate reads after specified length for R1 and R2 reads, respectively.
41 maxLen = 600, # discard reads longer than specified.
42 minLen = 100, # discard reads shorter than specified.
43 minQ = 2, # discard reads (after truncation) that contain a quality score below specified value.
44 matchIDs = TRUE, # output paired-end reads with matching IDs (for merging).
45 compress = TRUE, # gzip the output
46 multithread = TRUE) # use multiple threads
47 saveRDS(qfilt, file.path(path_results, "qfilt_reads.rds"))
48
49 # make sequence count report
50 seq_count = cbind(qfilt)
51 colnames(seq_count) = c("input", "qualFiltered")
52 seq_count = as.data.frame(seq_count)
53 seq_count$sample = sample_names
54 # reorder columns
55 seq_count = seq_count[, c("sample", "input", "qualFiltered")]
56 write.csv(seq_count, file.path(path_results, "seq_count_summary.csv"),
57 row.names = FALSE, quote = FALSE)
58
59 # save filtered R objects for denoising and merging (below)
60 filtR1 = sort(list.files(path_results, pattern = ".R1.fastq.gz", full.names = TRUE))
61 filtR2 = sort(list.files(path_results, pattern = ".R2.fastq.gz", full.names = TRUE))
62 sample_names = sapply(strsplit(basename(filtR1), ".R1.fastq.gz"), `[`, 1)
63 saveRDS(filtR1, file.path(path_results, "filtR1.rds"))
64 saveRDS(filtR2, file.path(path_results, "filtR2.rds"))
65 saveRDS(sample_names, file.path(path_results, "sample_names.rds"))
66 #-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#
67 #set working directory back to "/multiRunDir"
68 setwd(wd)
69 i = i + 1
70 }
71 }
Denoise and merge paired-end reads
1 #!/usr/bin/Rscript
2 ## workflow to perform DADA2 denoising and merging
3
4 # load dada2 library
5 library('dada2')
6
7 # capturing the directory structure when working with multiple runs
8 wd = getwd() # -> wd is "~/multiRunDir"
9 dirs = list.dirs(recursive = FALSE)
10 for (i in 1:length(dirs)) {
11 if(length(dirs) > 1) {
12 setwd(dirs[i])
13 print(paste0("Working with ", dirs[i]))
14 #-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#
15 #load quality filtered files
16 filtR1 = readRDS("qualFiltered_out/filtR1.rds")
17 filtR2 = readRDS("qualFiltered_out/filtR2.rds")
18 qfilt = readRDS("qualFiltered_out/qfilt_reads.rds")
19 sample_names = readRDS("qualFiltered_out/sample_names.rds")
20 cat("\n sample names = ", sample_names, "\n ")
21 names(filtR1) = sample_names
22 names(filtR2) = sample_names
23
24 # create output dir
25 path_results = "denoised_merged"
26 dir.create(path_results, showWarnings = FALSE)
27
28 print("# Denoising ...")
29 # learn the error rates
30 errF = learnErrors(filtR1, multithread = TRUE)
31 errR = learnErrors(filtR2, multithread = TRUE)
32
33 # make error rate figures
34 pdf(file.path(path_results, "Error_rates_R1.pdf"))
35 print( plotErrors(errF) )
36 dev.off()
37 pdf(file.path(path_results, "Error_rates_R2.pdf"))
38 print( plotErrors(errR) )
39 dev.off()
40
41 # Sample inference and merger of paired-end reads
42 mergers = vector("list", length(sample_names))
43 names(mergers) = sample_names
44 for(sample in sample_names) {
45 cat("\n -- Processing:", sample, "\n")
46 derepF = derepFastq(filtR1[[sample]])
47 ddF = dada(derepF, err = errF, multithread = TRUE)
48 derepR = derepFastq(filtR2[[sample]])
49 ddR = dada(derepR, err = errR, multithread = TRUE)
50 merger = mergePairs(ddF, derepF, ddR, derepR)
51 mergers[[sample]] = merger
52 }
53 rm(derepF); rm(derepR)
54 gc()
55 saveRDS(mergers, (file.path(path_results, "mergers.rds")))
56
57 # make sequence table
58 ASV_tab = makeSequenceTable(mergers)
59 #write RDS object
60 saveRDS(ASV_tab, (file.path(path_results, "rawASV_table.rds")))
61
62 # make sequence count report
63 getN = function(x) sum(getUniques(x))
64 #remove 0 seqs samples from qfilt statistics
65 row_sub = apply(qfilt, 1, function(row) all(row !=0 ))
66 qfilt = qfilt[row_sub, ]
67 seq_count = cbind(qfilt, sapply(mergers, getN))
68 colnames(seq_count) = c("input", "qualFiltered", "denoised_and_merged")
69 rownames(seq_count) = sample_names
70 write.csv(seq_count, file.path(path_results, "seq_count_summary.csv"),
71 row.names = TRUE, quote = FALSE)
72 #-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#
73 print("--------")
74 setwd(wd)
75 i = i + 1
76 }
77 }
Chimera filtering
1 #!/usr/bin/Rscript
2 ## workflow to perform chimera filtering within DADA2
3
4 # load libraries
5 library('dada2')
6 library('openssl')
7
8 # chimera filtering method
9 method = "consensus"
10
11 # collapse ASVs that have no mismatshes or internal indels (identical up to shifts and/or length)
12 collapseNoMismatch = "true" #true/false
13
14 # capturing the directory structure when working with multiple runs
15 wd = getwd() # -> wd is "~/multiRunDir"
16 dirs = list.dirs(recursive = FALSE)
17 for (i in 1:length(dirs)) {
18 if(length(dirs) > 1) {
19 setwd(dirs[i])
20 print(paste0("Working with ", dirs[i]))
21 #-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#
22 # load denoised and merged ASVs
23 rawASV_table = readRDS("denoised_merged/rawASV_table.rds")
24 # create output dir
25 path_results="ASV_table"
26 dir.create(path_results, showWarnings = FALSE)
27 # Remove chimeras
28 print("Removing chimeric ASVs ...")
29 chim_filt = removeBimeraDenovo(
30 rawASV_table, method = method,
31 multithread = TRUE,
32 verbose = TRUE)
33 saveRDS(chim_filt, "ASV_table/chim_filt.rds")
34
35 ### format and save ASV table and ASVs.fasta files
36 # sequence headers
37 asv_seqs = colnames(chim_filt)
38 asv_headers = openssl::sha1(asv_seqs)
39 # transpose sequence table
40 tchim_filt = t(chim_filt)
41 # add sequences to 1st column
42 tchim_filt = cbind(row.names(tchim_filt), tchim_filt)
43 colnames(tchim_filt)[1] = "Sequence"
44 # row names as sequence headers
45 row.names(tchim_filt) = asv_headers
46 # write ASVs.fasta to path_results
47 asv_fasta = c(rbind(paste(">", asv_headers, sep=""), asv_seqs))
48 write(asv_fasta, file.path(path_results, "ASVs.fasta"))
49 # write ASVs table to path_results
50 write.table(tchim_filt, file.path(path_results, "ASV_table.txt"),
51 sep = "\t", col.names = NA,
52 row.names = TRUE, quote = FALSE)
53
54 ### collapse ASVs that have no mismatshes or internal indels
55 # (identical up to shifts and/or length)
56 if (collapseNoMismatch == "true") {
57 print("Collapsing identical ASVs ...")
58 ASV_tab_collapsed = collapseNoMismatch(chim_filt,
59 minOverlap = 20, orderBy = "abundance",
60 identicalOnly = FALSE, vec = TRUE,
61 band = -1, verbose = TRUE)
62 saveRDS(ASV_tab_collapsed, file.path(path_results, "ASV_table_collapsed.rds"))
63
64 ### format and save ASV table and ASVs.fasta files
65 # sequence headers
66 asv_seqs = colnames(ASV_tab_collapsed)
67 asv_headers = openssl::sha1(asv_seqs)
68 # transpose sequence table
69 tASV_tab_collapsed = t(ASV_tab_collapsed)
70 # add sequences to 1st column
71 tASV_tab_collapsed = cbind(row.names(tASV_tab_collapsed), tASV_tab_collapsed)
72 colnames(tASV_tab_collapsed)[1] = "Sequence"
73 #row names as sequence headers
74 row.names(tASV_tab_collapsed) = asv_headers
75 # write ASVs.fasta to path_results
76 asv_fasta = c(rbind(paste(">", asv_headers, sep=""), asv_seqs))
77 write(asv_fasta, file.path(path_results, "ASVs_collapsed.fasta"))
78 # write ASVs table to path_results
79 write.table(tASV_tab_collapsed, file.path(path_results, "ASVs_table_collapsed.txt"),
80 sep = "\t", col.names = NA, row.names = TRUE, quote = FALSE)
81
82 # print summary
83 print(paste0("Output = ", length(colnames(ASV_tab_collapsed)),
84 " chimera filtered (+collapsed) ASVs, with a total of ",
85 sum(rowSums(ASV_tab_collapsed)),
86 " sequences."))
87 print("--------")
88 } else {
89 # print summary
90 print(paste0("Output = ", length(colnames(chim_filt)),
91 " chimera filtered ASVs, with a total of ",
92 sum(rowSums(chim_filt)),
93 " sequences."))
94 print("--------")
95 }
96 #-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#
97 setwd(wd)
98 i = i + 1
99 }
100 }
Remove tag-jumps
1#!/usr/bin/Rscript
2## Script to perform tag-jump removal; (C) Vladimir Mikryukov,
3 # edit, Sten Anslan
4
5 # load libraries
6 library(data.table)
7
8 # set parameters
9 set_f = 0.03 # f-parameter of UNCROSS (e.g., 0.03)
10 set_p = 1 # p-parameter (e.g., 1.0)
11
12 # output dir
13 path_results="ASV_table"
14
15 # capturing the directory structure when working with multiple runs
16 wd = getwd() # -> wd is "~/multiRunDir"
17 dirs = list.dirs(recursive = FALSE)
18 for (i in 1:length(dirs)) {
19 if(length(dirs) > 1) {
20 setwd(dirs[i])
21 print(paste0("Working with ", dirs[i]))
22 #-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#
23 # load ASV table
24 # loading ASV_table_collapsed if collapseNoMismatch was "true" (above)
25 if (file.exists("ASV_table/ASV_table_collapsed.rds") == TRUE) {
26 tab = readRDS("ASV_table/ASV_table_collapsed.rds")
27 cat("input table = ASV_table/ASV_table_collapsed.rds\n")
28 } else { # loading chimera filtered ASV table
29 tab = readRDS("ASV_table/chim_filt.rds")
30 cat("input table = ASV_table/chim_filt.rds\n")
31 }
32
33 # format ASV table
34 ASVTABW = as.data.table(t(tab), keep.rownames = TRUE)
35 colnames(ASVTABW)[1] = "ASV"
36 # convert to long format
37 ASVTAB = melt(data = ASVTABW, id.vars = "ASV",
38 variable.name = "SampleID", value.name = "Abundance")
39 # remove zero-OTUs
40 ASVTAB = ASVTAB[ Abundance > 0 ]
41 # estimate total abundance of sequence per plate
42 ASVTAB[ , Total := sum(Abundance, na.rm = TRUE), by = "ASV" ]
43
44 ## UNCROSS score
45 uncross_score = function(x, N, n, f = 0.01, tmin = 0.1, p = 1){
46 z = f * N / n # Expected treshold
47 sc = 2 / (1 + exp(x/z)^p) # t-score
48 res = data.table(Score = sc, TagJump = sc >= tmin)
49 return(res)
50 }
51
52 # esimate UNCROSS score
53 cat(" estimating UNCROSS score\n")
54 ASVTAB = cbind(
55 ASVTAB,
56 uncross_score(
57 x = ASVTAB$Abundance,
58 N = ASVTAB$Total,
59 n = length(unique(ASVTAB$SampleID)),
60 f = as.numeric(set_f),
61 p = as.numeric(set_p)
62 )
63 )
64 cat(" number of tag-jumps: ", sum(ASVTAB$TagJump, na.rm = TRUE), "\n")
65
66 # tag-jump stats
67 TJ = data.table(
68 Total_reads = sum(ASVTAB$Abundance),
69 Number_of_TagJump_Events = sum(ASVTAB$TagJump),
70 TagJump_reads = sum(ASVTAB[ TagJump == TRUE ]$Abundance, na.rm = T))
71
72 TJ$ReadPercent_removed = with(TJ, (TagJump_reads / Total_reads * 100))
73 fwrite(x = TJ, file = "ASV_table/TagJump_stats.txt", sep = "\t")
74
75 # prepare ASV tables, remove tag-jumps
76 ASVTAB = ASVTAB[ TagJump == FALSE ]
77 # convert to wide format
78 RES = dcast(data = ASVTAB,
79 formula = ASV ~ SampleID,
80 value.var = "Abundance", fill = 0)
81 # sort rows (by total abundance)
82 clz = colnames(RES)[-1]
83 otu_sums = rowSums(RES[, ..clz], na.rm = TRUE)
84 RES = RES[ order(otu_sums, decreasing = TRUE) ]
85
86 # output table that is compadible with dada2
87 output = as.matrix(RES, sep = "\t", header = TRUE, rownames = 1,
88 check.names = FALSE, quote = FALSE)
89 output = t(output)
90 saveRDS(output, ("ASV_table/ASV_table_TagJumpFiltered.rds"))
91
92 ### format and save ASV table and ASVs.fasta files
93 # sequence headers
94 asv_seqs = colnames(output)
95 asv_headers = openssl::sha1(asv_seqs)
96 # transpose sequence table
97 toutput = t(output)
98 # add sequences to 1st column
99 toutput = cbind(row.names(toutput), toutput)
100 colnames(toutput)[1] = "Sequence"
101 #row names as sequence headers
102 row.names(toutput) = asv_headers
103 # write ASVs.fasta to path_results
104 asv_fasta = c(rbind(paste(">", asv_headers, sep=""), asv_seqs))
105 write(asv_fasta, file.path(path_results, "ASVs_TagJumpFiltered.fasta"))
106 # write ASVs table to path_results
107 write.table(toutput, file.path(path_results, "ASV_table_TagJumpFiltered.txt"),
108 sep = "\t", col.names = NA, row.names = TRUE, quote = FALSE)
109
110 # print summary
111 print(paste0("Output = ", length(colnames(output)), " ASVs, with a total of ",
112 sum(rowSums(output)), " sequences."))
113
114 #-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#-#
115 print("--------")
116 setwd(wd)
117 i = i + 1
118 }
119 }
Merge sequencing runs
1 #!/usr/bin/Rscript
2 ## Merge sequencing runs, if working with multiple ones
3
4 # load libraries
5 library('dada2')
6
7 # after merging multiple ASV tables ...
8 # collapse ASVs that have no mismatshes or internal indels
9 collapseNoMismatch = "true" #true/false
10
11 # capturing the directory structure when working with multiple runs
12 wd = getwd() # -> wd is "~/multiRunDir"
13 dirs = list.dirs(recursive = FALSE)
14 tables = c()
15 # load tables from multiple sequencing runs (dirs)
16 for (i in 1:length(dirs)) {
17 if(length(dirs) > 1) {
18 setwd(dirs[i])
19 tables = append(tables, print(file.path(paste0(dirs[i], "/ASV_table"),
20 "ASV_table_TagJumpFiltered.rds")))
21 setwd(wd)
22 i = i + 1
23 }
24 }
25
26 # Merge multiple ASV tables
27 print("# Merging multiple ASV tables ...")
28 ASV_tables = lapply(tables, readRDS)
29 merged_table = mergeSequenceTables(tables = ASV_tables, repeats = "sum", tryRC = FALSE)
30
31 ### collapse ASVs that have no mismatshes or internal indels
32 if (collapseNoMismatch == "true") {
33 print("# Collapsing identical ASVs ...")
34 merged_table_collapsed = collapseNoMismatch(merged_table,
35 minOverlap = 20, orderBy = "abundance",
36 identicalOnly = FALSE, vec = TRUE,
37 band = -1, verbose = TRUE)
38 saveRDS(merged_table_collapsed, "merged_table_collapsed.rds")
39
40 ### format and save ASV table and ASVs.fasta files
41 # sequence headers
42 asv_seqs = colnames(merged_table_collapsed)
43 asv_headers = openssl::sha1(asv_seqs)
44 # transpose sequence table
45 tmerged_table_collapsed = t(merged_table_collapsed)
46 # add sequences to 1st column
47 tmerged_table_collapsed = cbind(row.names(tmerged_table_collapsed), tmerged_table_collapsed)
48 colnames(tmerged_table_collapsed)[1] = "Sequence"
49 #row names as sequence headers
50 row.names(tmerged_table_collapsed) = asv_headers
51 # write ASVs.fasta
52 asv_fasta = c(rbind(paste(">", asv_headers, sep=""), asv_seqs))
53 write(asv_fasta, "ASVs.merged_collapsed.fasta")
54 # write ASVs table
55 write.table(tmerged_table_collapsed, "ASV_table.merged_collapsed.txt",
56 sep = "\t", col.names = NA, row.names = TRUE, quote = FALSE)
57
58 # print summary
59 print(paste0("Output = ", length(colnames(merged_table_collapsed)),
60 " ASVs, with a total of ",
61 sum(rowSums(merged_table_collapsed)),
62 " sequences."))
63 } else {
64 saveRDS(merged_table, "merged_table.rds")
65 ### format and save ASV table and ASVs.fasta files
66 # sequence headers
67 asv_seqs = colnames(merged_table)
68 asv_headers = openssl::sha1(asv_seqs)
69 # transpose sequence table
70 tmerged_table = t(merged_table)
71 # add sequences to 1st column
72 tmerged_table = cbind(row.names(tmerged_table), tmerged_table)
73 colnames(tmerged_table)[1] = "Sequence"
74 #row names as sequence headers
75 row.names(tmerged_table) = asv_headers
76 # write ASVs.fasta to path_results
77 asv_fasta = c(rbind(paste(">", asv_headers, sep=""), asv_seqs))
78 write(asv_fasta, "ASVs.merged.fasta")
79 # write ASVs table to path_results
80 write.table(tmerged_table, "ASV_table.merged.txt",
81 sep = "\t", col.names = NA, row.names = TRUE, quote = FALSE)
82
83 # print summary
84 print(paste0("Output = ", length(colnames(merged_table)),
85 " ASVs, with a total of ",
86 sum(rowSums(merged_table)),
87 " sequences."))
88 }
Extract ITS1/ITS2 region
Extract the ITS1/ITS2 region, i.e., clip conservative primer binding sites (18S, 5.8S, 28S) from the ASVs, for making ASVs that differ only withing the ITS1/ITS2 part.
1 #!/bin/bash
2
3 # specify the input ASVs fasta file
4 fasta="ASVs_TagJumpFiltered.fasta" # ASVs_TagJumpFiltered.fasta/ASVs.merged.fasta
5 # specify the target ITS region
6 region="ITS2" # ITS1/ITS2
7
8 # output dir
9 output_dir=$"ITSx_out"
10 mkdir $output_dir
11
12 # ITSx
13 ITSx -i $fasta \
14 --save_regions $region \
15 -o $output_dir/${fasta%.*} \
16 --preserve T \
17 --graphical F \
18 -t all \
19 --cpu 20 \
20 -E 0.01 \
21 --complement F \
22 --truncate T
1 #!/bin/bash
2
3 vsearch --derep_fulllength $output_dir/${fasta%.*}.$region.${fasta##*.} \
4 --output $output_dir/${fasta%.*}.$region.derep.${fasta##*.} \
5 --uc $output_dir/derep.uc \
6 --fasta_width 0
7
8 # export vars for R
9 derep_fasta="$output_dir/${fasta%.*}.$region.derep.${fasta##*.}"
10 export derep_fasta
11 export output_dir # output_dir=$"ITSx_out"
1 #!/usr/bin/Rscript
2
3 ### Generate ASV table based on the dereplicated ASVs that went through ITSx
4 ### Herein "OTUs" refer to "ASVs after ITSx".
5
6 # specify input ASV table (the same one as for 'get the size of ASVs')
7 ASV_table="ASV_table_TagJumpFiltered.fasta"
8
9 # read output dir
10 output_dir = Sys.getenv('output_dir')
11
12 # read output from vsearch clustering (-uc OTU.uc)
13 inp_UC = file.path(output_dir, "derep.uc")
14 ################################
15 library(data.table)
16 # load input data - ASV table
17 ASV_table = fread(file = ASV_table, header = TRUE, sep = "\t")
18
19 ## Load input data - UC mapping file
20 UC = fread(file = inp_UC, header = FALSE, sep = "\t")
21 UC = UC[ V1 != "S" ]
22 UC[, ASV := tstrsplit(V9, ";", keep = 1) ]
23 UC[, OTU := tstrsplit(V10, ";", keep = 1) ]
24 UC[V1 == "C", OTU := ASV ]
25 UC = UC[, .(ASV, OTU)]
26
27 # convert ASV table to long format
28 ASV = melt(data = ASV_table,
29 id.vars = colnames(ASV_table)[1],
30 variable.name = "SampleID", value.name = "Abundance")
31 ASV = ASV[ Abundance > 0 ]
32 # add colnames, to make sure 1st is 'ASV'
33 colnames(ASV) = c("ASV", "SampleID", "Abundance")
34
35 # add OTU IDs
36 ASV = merge(x = ASV, y = UC, by = "ASV", all.x = TRUE)
37 # summarize
38 OTU = ASV[ , .(Abundance = sum(Abundance, na.rm = TRUE)),
39 by = c("SampleID", "OTU")]
40
41 # reshape OTU table to wide format
42 OTU_table = dcast(data = ASV,
43 formula = OTU ~ SampleID,
44 value.var = "Abundance",
45 fun.aggregate = sum, fill = 0)
46
47 # write OTU table
48 # OTU names correspond to most abundant ASV in an OTU
49 fwrite(x = OTU_table, file = file.path(output_dir,
50 "ASV_table_ITSx.txt"), sep = "\t")
1 #!/bin/bash
2
3 # make fasta file corresponding to ASV table
4 awk 'NR>1{print $1}' ASV_table_ITSx.txt > ASVs.list
5 cat $fasta | seqkit grep -w 0 -f ASVs.list > ASVs.ITSx.fasta
Taxonomy assignment
Taxonomy assignment with the SINTAX against EUKARYOME database .
— Download the EUKARYOME (v1.9.2) for SINTAX here (click) —
1 #!/bin/bash
2
3 # download the EUKARYOME reference databse
4 wget \
5 "https://sisu.ut.ee/wp-content/uploads/sites/643/SINTAX_EUK_ITS_v1.9.zip"
6 # unzip the database and edit name
7 unzip SINTAX_EUK_ITS_v1.9.zip -d EUKARYOME1.9.2_sintax
8
9 # specify reference database
10 reference_database="EUKARYOME1.9.2_sintax/SINTAX_EUK_ITS_v1.9.fasta"
11 reference_database=$(realpath $reference_database) # get database names with full path
12
13 # specify input fasta file
14 cd ASV_table
15 ASV_fasta="ASVs.ITSx.fasta"
16
17 # Run SINTAX classifier
18 vsearch -sintax $ASV_fasta \
19 -db $reference_database \
20 -tabbedout taxonomy.txt \
21 -strand both \
22 -sintax_cutoff 0.8 \
23 --threads 8
Clustering ASVs to OTUs
1 #!/usr/bin/env Rscript
2
3 # specify input ASVs table and fasta
4 ASV_table="ASV_table_ITSx.txt" # specify ASV table file
5 ASV_fasta="ASVs.ITSx.fasta" # specify ASVs fasta file
6
7 ################################
8 library(Biostrings)
9 # Read the ASV table
10 ASV_table = read.table(ASV_table, sep = "\t", check.names = F,
11 header = T, row.names = 1)
12
13 # add 'sum' column
14 ASV_table$sum = rowSums(ASV_table)
15 # make ASV_sums object
16 ASV_sums = setNames(ASV_table$sum, rownames(ASV_table))
17
18 # Read the FASTA file
19 ASV_fasta = readDNAStringSet(ASV_fasta)
20
21 # add ";size=*" to ASV_fasta
22 names(ASV_fasta) = sapply(names(ASV_fasta), function(header) {
23 paste0(header, ";size=", ASV_sums[header])
24 })
25 # write fasta file
26 writeXStringSet(ASV_fasta, "ASVs.size.fasta",
27 width = max(width(ASV_fasta)))
1 #!/bin/bash
2
3 # specify the clustering threshold
4 clustering_thresh="0.97"
5
6 # make output dir.
7 output_dir="OTU_table"
8 mkdir -p $output_dir
9 export output_dir
10
11 ### cluster ASVs using vsearch.
12 vsearch --cluster_fast ASVs.size.fasta \
13 --id $clustering_thresh \
14 --iddef 2 \
15 --sizein \
16 --xsize \
17 --fasta_width 0 \
18 --centroids $output_dir/OTUs.fasta \
19 --uc $output_dir/OTUs.uc
1 #!/usr/bin/Rscript
2
3 # specify input ASV table (the same one as for 'get the size of ASVs')
4 ASV_table="ASV_table_ITSx.txt"
5
6 # read output dir
7 output_dir = Sys.getenv('output_dir')
8
9 # read output from vsearch clustering (-uc OTU.uc)
10 inp_UC = file.path(output_dir, "OTUs.uc")
11 ################################
12 library(data.table)
13 # load input data - ASV table
14 ASV_table = fread(file = ASV_table, header = TRUE, sep = "\t")
15
16 ## Load input data - UC mapping file
17 UC = fread(file = inp_UC, header = FALSE, sep = "\t")
18 UC = UC[ V1 != "S" ]
19 UC[, ASV := tstrsplit(V9, ";", keep = 1) ]
20 UC[, OTU := tstrsplit(V10, ";", keep = 1) ]
21 UC[V1 == "C", OTU := ASV ]
22 UC = UC[, .(ASV, OTU)]
23
24 # convert ASV table to long format
25 ASV = melt(data = ASV_table,
26 id.vars = colnames(ASV_table)[1],
27 variable.name = "SampleID", value.name = "Abundance")
28 ASV = ASV[ Abundance > 0 ]
29 # add colnames, to make sure 1st is 'ASV'
30 colnames(ASV) = c("ASV", "SampleID", "Abundance")
31
32 # add OTU IDs
33 ASV = merge(x = ASV, y = UC, by = "ASV", all.x = TRUE)
34 # summarize
35 OTU = ASV[ , .(Abundance = sum(Abundance, na.rm = TRUE)),
36 by = c("SampleID", "OTU")]
37
38 # reshape OTU table to wide format
39 OTU_table = dcast(data = ASV,
40 formula = OTU ~ SampleID,
41 value.var = "Abundance",
42 fun.aggregate = sum, fill = 0)
43
44 # write OTU table
45 # OTU names correspond to most abundant ASV in an OTU
46 fwrite(x = OTU_table, file = file.path(output_dir,
47 "OTU_table.txt"), sep = "\t")
Post-clustering
Post-cluster OTUs with LULU to merge consistently co-occurring ‘daughter-OTUs’.
1 #!/bin/bash
2
3 # go to directrory that contains OTUs
4 cd $output_dir # 'OTU_table' in this case
5
6 # make blast database for post-clustering
7 makeblastdb -in OTUs.fasta -parse_seqids -dbtype nucl
8
9 # generate match list for post-clustering
10 blastn -db OTUs.fasta \
11 -outfmt '6 qseqid sseqid pident' \
12 -out match_list.txt \
13 -qcov_hsp_perc 75 \
14 -perc_identity 90 \
15 -query OTUs.fasta \
16 -num_threads 20
1 #!/usr/bin/Rscript
2
3 # specify minimum threshold of sequence similarity considering any OTU as an error of another
4 min_match = "90"
5
6 # specify OTU table
7 OTU_table="OTU_table.txt"
8
9 ################################
10 library(devtools)
11 # load OTU table and match list
12 otutable = read.table(OTU_table, header = T, row.names = 1, sep = "\t")
13 matchlist = read.table("match_list.txt")
14
15 curated_result = lulu::lulu(otutable, matchlist,
16 minimum_match = min_match)
17
18 # write post-clustered OTU table to file
19 curated_table = curated_result$curated_table
20 curated_table = cbind(OTU = rownames(curated_table), curated_table)
21 write.table(curated_table, file ="OTU_table_LULU.txt",
22 sep = "\t", row.names = F, quote = FALSE)
23 write.table(curated_result$discarded_otus,
24 file ="merged_units.lulu", col.names = FALSE, quote = FALSE)
Note
Note that if the sample names start with a number, then the output OTU table may contain “X” prefix in the sample names.
1 #!/bin/bash
2
3 # specify post-clustered table
4 OTU_table="OTU_table_LULU.txt"
5 # specify pre post-clustered OTUs fasta file
6 OTUs_fasta="OTUs.fasta"
7
8 # get matching OTUs
9 awk 'NR>1{print $1}' $OTU_table > OTUs_LULU.list
10 cat $OTUs_fasta | \
11 seqkit grep -w 0 -f OTUs_LULU.list > OTUs_LULU.fasta
12
13 # get matching SINTAX taxonomy results
14 head -n 1 ../taxonomy.txt > taxonomy.txt
15 cat ../taxonomy.txt | \
16 grep -wf OTUs_LULU.list >> taxonomy.txt
17
18 # remove unnecessary files
19 rm OTUs.fasta.n*
20
21 # move OTU_table two directories down
22 cd ..
23 mv $output_dir ../..
Note
The final OTUs data is OTU_table_LULU.txt and OTUs_LULU.fasta in the OTU_table directory.
The matching SINTAX taxonomy files are taxonomy.txt in the OTU_table directory.
We acknowledge CSC - IT Center for Science, Finland, for computational resources while building and testing this workflows.
![]()

![]()
![]()