![]()
ITS2
Amplicon library preparation for nuclear ribosomal internal transcribed spacer 2 (ITS2) region to target fungal communities using 2-step PCR with primers gITS7ngs (GTGARTCATCRARTYTTTG) and ITS4ngsUni (CCTSCSCTTANTDATATGC) [Tedersoo & Lindahl 2016].
Herein processes follow lab SOP for the ‘Characterization of Prokaryotic and Eukaryotic Biodiversity from Soil Samples’ (Chaves et al., 2025); the workflow is hosted in WorkflowHub (hosts the downloadable PDF).
Besides used primers and the PCR conditions for the 1st PCR, the protocol in identical to COI and 16S library prep.
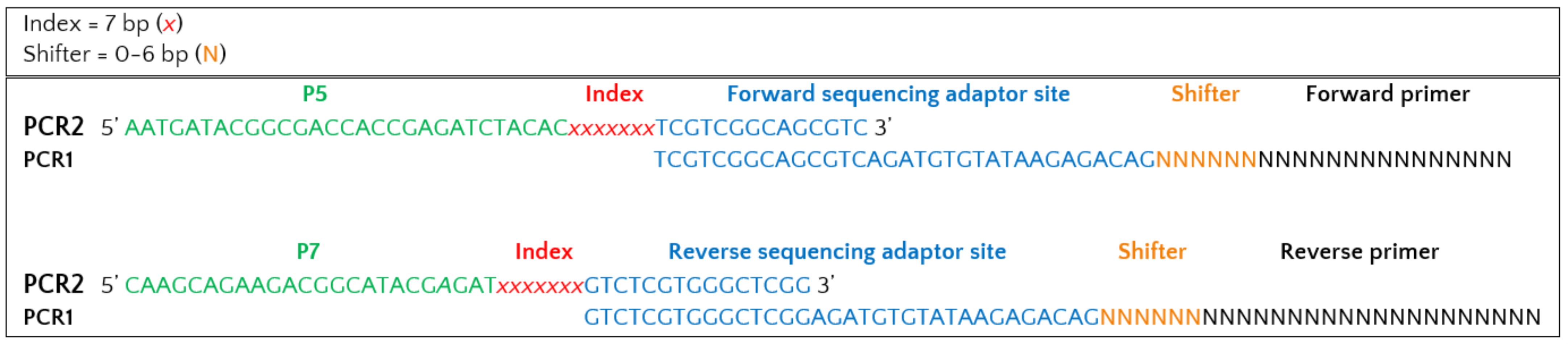
Primer constructs for 1st PCR
Forward sequencing adaptor site |
Shifter* |
Forward primer |
|---|---|---|
TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG |
NNNNNN |
GTGARTCATCRARTYTTTG |
Reverse sequencing adaptor site |
Shifter* |
Reverse primer |
|---|---|---|
GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG |
NNNNN |
CCTSCSCTTANTDATATGC |
* Shifter is 0-6 bp
Indexing primer constructs for 2nd PCR (indexing PCR) with overhangs to sequencing adaptor sites:
P5 (forward) adapter |
Index |
Forward overhang |
|---|---|---|
AATGATACGGCGACCACCGAGATCTACAC |
NNNNNNN |
TCGTCGGCAGCGTC |
P7 (reverse) adapter |
Index |
Reverse overhang |
|---|---|---|
CAAGCAGAAGACGGCATACGAGAT |
NNNNNNN |
GTCTCGTGGGCTCGG |
Full construct:
1st PCR
Consumables:
Items |
Initial Concentration |
Quantity |
Storage |
|---|---|---|---|
eDNA (dilution plate) |
10 ng/µL |
2 µL per sample |
-15° to -25°C |
Amplicon PCR Forward Primer |
10 µM |
0.25 µL per sample |
-15° to -25°C |
Amplicon PCR Reverse Primer |
10 µM |
0.25 µL per sample |
-15° to -25°C |
Qiagen Master Mix |
5x |
5 µL per sample |
-15° to -25°C |
Ultrapure water |
2.5 µL per sample |
||
96-well 0.2 mL PCR plate |
1 plate |
Total volume per PCR reaction = 10µL.
PCR conditions:
95ºC for 15 minutes |
|
95ºC for 30 seconds
58ºC for 30 seconds
72ºC for 60 seconds
|
35 cycles
|
72ºC for 10 minutes |
|
Hold at 10ºC |
Expected amplicon size = 499 bp (insert length (~300~500 bp) + primer lengths + 6 bp NNs (avg.) + 67 bp overhangs).
● Test the PCR success of all samples through electrophoresis of 2 µL using 2% agarose gel.
● Dilute samples 1:4 using ultrapure water.
2nd PCR (indexing PCR)
Consumables:
Items
|
Initial
Concentration
|
Quantity
|
Storage
|
PCR 1 (diluted 1:4) |
n.i. |
2.8 µL per sample |
-15° to -25°C |
P5-P7 Index Primer Mix |
10 µM |
1.4 µL per sample |
-15° to -25°C |
KAPA HiFi Hot-Start Ready Mix |
2x |
7 µL per sample |
-15° to -25°C |
H2O |
2.8 µL per sample |
||
96-well 0.2 mL PCR plate |
1 plate |
Procedure:
Transfer 1.4 µL of each mixed combination of P5 and P7 indexing primers to a new plate.
Set up the following reaction per sample:
1X |
|
|---|---|
KAPA HiFi Hot-Start Ready Mix |
7 µL |
H2O |
2.8 µL |
Total |
9.8 µL |
Mix the reagents by pipetting, spin down and distribute it in each well.
Add 2.8 µL of the diluted PCR1 product.
Seal plate and perform PCR in a thermal cycler using the following conditions:
95ºC for 3 minutes |
|
95ºC for 30 seconds
55ºC for 30 seconds
72ºC for 30 seconds
|
8 cycles
|
72ºC for 5 minutes |
|
Hold at 10ºC |
Test size shift between PCR1 and PCR2 amplicons of 15% of samples (e.g. 4 sets of 4 samples selected from random rows) through electrophoresis in 2% agarose gel.
Clean PCR products
This step uses magnetic beads to purify PCR products from free primers and primer-dimers.
Equipment and consumables:
Items |
Quantity |
Storage |
|---|---|---|
Qiagen EB Buffer |
25 µL per sample |
15ºC - 25ºC |
KAPA HyperPure Beads |
8 µL per sample |
4ºC |
Freshly prepared 80% ethanol (EtOH) |
300 µL per sample |
|
Cell culture plate (new) |
4 plates |
|
96-well PCR plate Non-skirted (VWR) |
1 plate |
|
Reservoirs |
1 |
|
Magnetic Bead Extractor for 96 Well Microplates (V&P Scientific) |
1 |
|
Low-bind microplate (Optional) |
1 |
Preparation:
Bring the KAPA HyperPure beads to room temperature for 30min prior to usage;
Prepare fresh 80% ethanol;
Prepare a 50mL tube with EB Buffer and protect from any direct light source;
Short spin the Amplicon PCR plate to collect condensation;
Clean the working space and material with disinfectant and ethanol;
Sterilize, under UV light for about 15min, four U-bottom 96-well plates, a falcon with freshly prepared 80% ethanol and EB Buffer.
Procedure:
Distribute the appropriate volume of beads in one of the U-bottom 96-well plates (U-plate 1).
Note: The volume of beads may depend on the ratio chosen, which varies according to library size. A standard ratio of 0,8x is used, adding 8 µL of beads for 10 µL of sample.
Transfer the full PCR volume (10 µL) into the plate containing the KAPA HyperPure Beads, carefully pipetting the entire volume up and down 10 times.
Incubate at room temperature without shaking for 3 min.
While in waiting, prepare three more U-bottom 96-well plates as following: two plates with 150µL 80% ethanol and one plate with 25µL EB Buffer.
Gently place a 96-well PCR plate on the plate from step 2 and attach the magnetic bead separation extractor for 2min or until the supernatant is cleared.
Carefully remove the extractor and submerge the beads into one of the plates with freshly prepared 80% ethanol (U-plate 2) for 30s.
Carefully remove the extractor and perform a second ethanol wash (U-plate 3).
Allow the beads to air-dry for 6-7min.
Note: Do not over-dry the beads, if they start to appear cracked immediately proceed to the next step.
Carefully immerse the beads into the EB buffer (U-plate 4) and release the PCR plate from the extractor.
Carefully resuspend the beads in EB buffer.
Attach the magnetic extractor to the PCR plate for 2min or until the supernatant is cleared.
Carefully remove the magnetic extractor and seal the U-bottom plate (or transfer it to a new low-bind PCR plate).
Pooling & quantification
Consumables:
Items |
|---|
Qiagen EB Buffer |
KAPA HyperPure Beads |
96-well 0.2 mL PCR plate |
KAPA Library Quantification Kit (Roche) |
Tapestation High Sensitivity D5000 (Agilent) |
Qubit HS (Themo Fisher Scientific) |
Procedure:
Quantify each library using spectrophotometry (e.g Nanodrop) to estimate average library concentration (ng/µL).
Pool libraries equimolarly at 50 ng by taking the corresponding uL from each library. The negative controls should be added at a maximum volume than any other single library (up to 20 µL).
Note: In cases where the sample does not have volume to take 50ng, use the smallest common concentration available
Clean the pool with KAPA HyperPure Beads.
Note: The volume of beads may depend on the ratio chosen, which varies according to library size. A ratio of 0,7x can be used, adding 70 µL of beads for 100 µL of sample.
Quantify library pool using KAPA Library Quantification Kit for qPCR, Qubit or Tapestation.
Dilute each library pool using Buffer EB according to specifications by sequencing provider (if needed).
Verify the final concentration of a library pool using KAPA Library Quantification Kit in qPCR.
References
Chaves, C., Najera Cortazar, L. A., Martins, F., Anslan, S., Beja-Pereira, A., Magalhães, M., & Price, B. (2025). Characterization of Prokaryotic and Eukaryotic Biodiversity from Soil Samples. WorkflowHub. https://doi.org/10.48546/workflowhub.sop.12.2
![]()

![]()
![]()